RTI 还报告指出,申办人并不总是随申请一起提交患者体验数据。在 92 份提交中,有 72 份包含患者体验数据,20 份不包含。在申办人未提交患者体验数据的三种情况下,审评人员评估了在其它地方获得的患者体验数据。研究结果显示,如果申办人不提供任何数据,FDA 将会从其它地方找到一些患者体验数据。Gnanasakthy 表示,“如果你是申办人,让 FDA 去别处找数据不是一个好主意。最好主动提交 FDA 将考虑的数据。但是我认为随着时间的推移,这种情况会有所改善。”
FDA 药品审评与研究中心(CDER)关注患者的药物开发主任 Robyn Bent 在会上表示,FDA 审评人员“在审评时会尽量做尽职调查以真正了解更大的图景。”她解释指出,“大量患者体验数据是作为申请过程的一部分交到 FDA,但我们也确实从其他人那里收到一些信息,而且很多时候我们的审评员也会做文献审评。”他们“还会参加外部主办的关注患者的药品开发会议以及 FDA 听证会”。
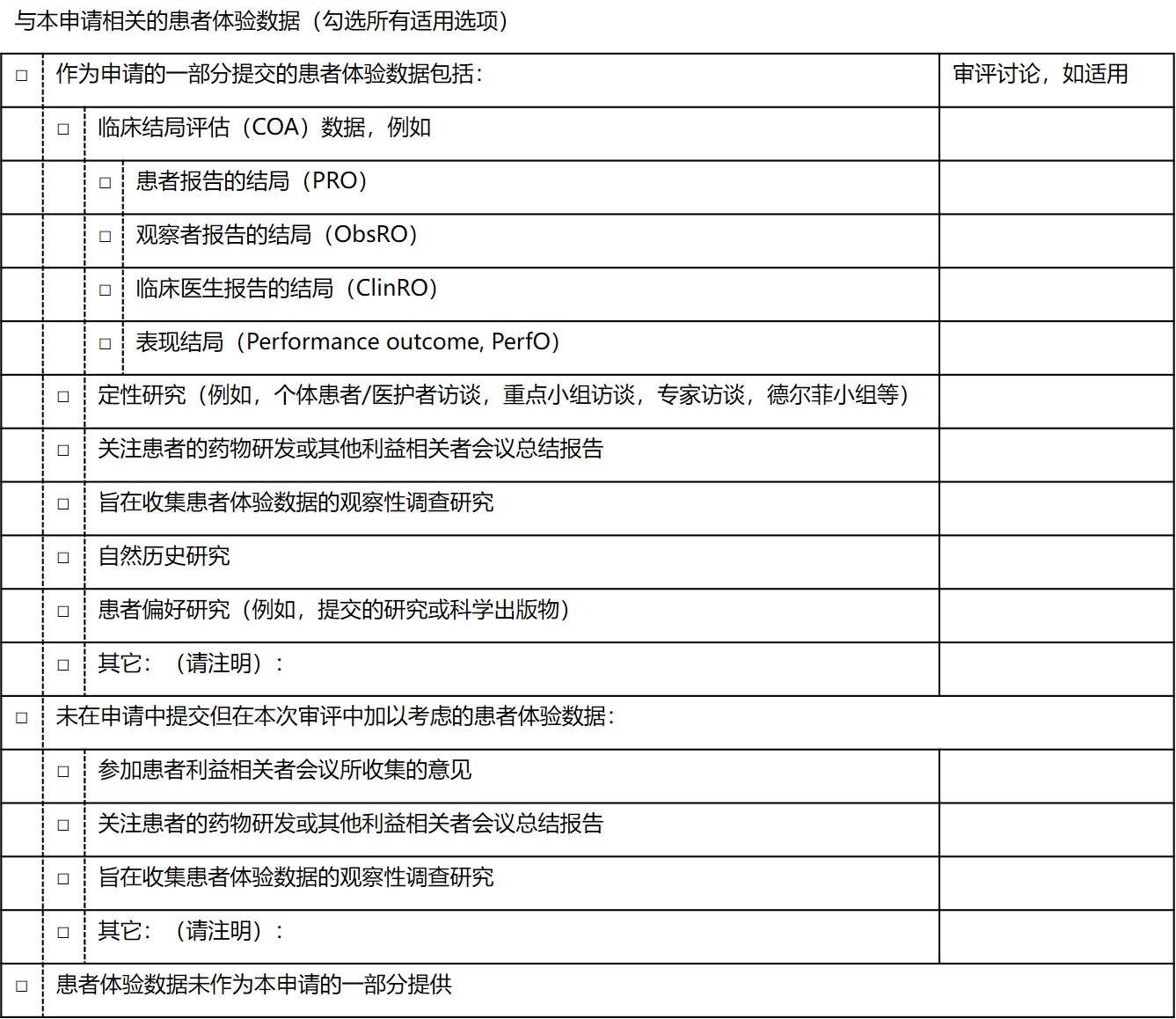
Bent 承认,FDA 最近发布了一份模板,供申办人在提交患者体验数据时使用,但可能并未充分提醒企业有关该工具的信息。该模板与患者体验数据表相似,在 2019 年 12 月的《电子通用技术文件(eCTD)技术符合性指南》中发布。
Bent 解释指出,“我们提供了有关 FDA 希望如何将患者体验数据作为申请的一部分提交的信息,因为这样在某种程度上我们可以一目了然地了解我们要寻找的内容,而如果申请人告诉我们去哪里寻找特定信息确实可以让我们的工作更轻松一点。”她指出,FDA 要求申办人提交一张表格,以作为审评人员对指南导航的一部分,识别和定位患者体验信息。