|
首页
>
资讯
>
FDA 用 AEMS 整合 FAERS 等全部7个不良反应报告平台
出自识林
FDA 用 AEMS 整合 FAERS 等全部7个不良反应报告平台
2026-03-18
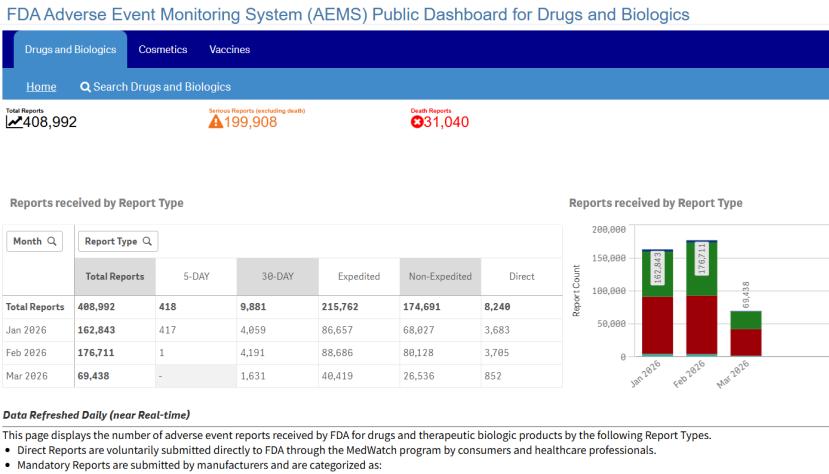
3月11日,FDA宣布启动新的不良事件监测系统(Adverse Event Monitoring System,AEMS)。该平台将整合FDA原有的七个不良反应报告数据库,解决此前系统分散、数据访问困难的问题。这是FDA局长Marty Makary“激进透明”政策在2025年公开完全回应函(CRL)和实时公开不良反应报告数据之后的又一举措。
Makary称原有系统已经过时且分散,导致重要数据难以获取。这些系统每年耗费约3700万美元的纳税人资金运营,并造成上市后监测中的“盲区”。新系统AEMS将作为一个统一的、直观的平台,为FDA科学家、研究人员和公众提供服务。
此前,FDA每年处理约600万份不良反应报告,这些报告分布在七个数据库中,包括药品和生物制品相关的FDA不良事件报告系统(FAERS)、与疾控中心共同管理的疫苗不良事件报告系统(VAERS)、含动物药品和动物食品报告两个数据库的不良事件报告系统(AERS)、医疗器械相关的制造商和使用者设施器械经验数据库(MAUDE)、人类食品投诉系统(HFCS)以及烟草产品的不良事件报告系统(CTPAE)。其中MAUDE、HFCS和CTPAE将在2026年5月完成整合。
在AEMS框架下,上述多种产品的不良反应报告将通过单一整合的仪表板展示(见题图)。根据FDA公告,截至2026年5月底,AEMS将实时公开所有FDA监管产品的不良反应报告数据。届时,FDA将完成历史不良反应数据的迁移,停用部分传统系统,并推出增强版应用程序接口(API)和数据分析工具。
FDA预计,AEMS的运行效率将帮助该机构在未来五年内节省约1.2亿美元。同时,由于AEMS实现报告的实时发布而非季度发布,预计将显著减少针对未发布不良反应报告的《信息自由法》(FOIA)请求。
FDA首席人工智能(AI)官Jeremy Walsh将此次系统整合描述为“机构历史上最大的技术变革......这是新FDA”。值得注意的是,首席AI官为AEMS背书,或体现FDA将AI用于药物警戒领域的积极态度。
然而局长的“激进透明”(Radical Transparency)政策可能是一把双刃剑。在缺乏专业解读的情况下,实时涌入的原始数据可能被社会误读,引发公众恐慌。例如对于普通用户而言,看到红色字体的死亡报告与某个药物挂钩时,很难保持理性态度。因此FDA强调该平台的数据局限性:
识林-实木
识林®版权所有,未经许可不得转载
【文件概要】
本文探讨FDA新药办公室(OND)主任Mary Thanh Hai关于公开完全回应函(CRL)及行动资料包的潜在政策动向。现行CRL披露机制中,产品质量和生产缺陷信息通常被删减,临床缺陷细节多保留,但CRL内容有限,仅反映监管结论而非完整审评逻辑。Thanh Hai提出,公开未批准药品的行动资料包可能更具价值,该资料包包含各审评部门的详细报告、监管摘要及决策备忘录,但实施面临资源消耗和商业机密泄露风险。她建议优先试点有效性补充申请的资料包公开,因其涉及已上市药品的超说明书用药场景,可为医疗决策提供参考。此举与FDA局长Makary推行的“激进透明”政策一致,目前CRL已逐步公开,但行动资料包披露仍处讨论阶段。 【适用范围】
本文适用于美国市场的新药和生物制品注册申请(含创新药及有效性补充申请),涉及企业包括跨国药企、Biotech及CRO/CDMO。 【影响评估】
若FDA扩大行动资料包公开范围,企业需加强CRL缺陷整改及审评策略预判能力,同时应对潜在商业信息泄露风险。透明度提升可能加速同类产品研发竞争,但亦有助于优化临床开发路径。 【实施建议】 - 必读岗位:注册、临床、研发
- 注册:跟踪FDA政策动态,评估CRL及行动资料包公开对申报策略的影响。
- 临床:分析公开资料包中的审评意见,优化临床试验设计。
- 研发:参考同类产品审评缺陷,规避技术风险。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |