|
首页
>
资讯
>
【周末杂谈】规章的起草说明
出自识林
2020-09-27
起草说明详述了FDA规章的来龙去脉和斟酌思考
这周三,FDA发布了两项拟议修订规章(21 CFR 201和801),澄清FDA是如何看待,因医生标签外用药或器械,企业是否要相应地修改产品标签【FDA 发布拟议法规澄清与标签外使用判定相关的证据】。这是个重要问题。众所周知,美国医生标签外用药,无需FDA批准,也无需告知企业。如果是纯粹的医生个人行为,非企业意向(intention),让企业修改标签不合适。但若存在企业有意地支持,则企业违法,可遭受重罚。
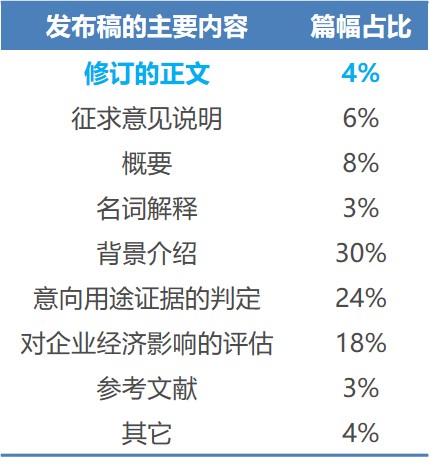
这两项修订内容,总共才半页纸,但其在联邦公告(Federal Register)上的发布,却长达11页纸。粗略统计了一下,发现各部分的篇幅占比如下,其中正文以外部分统称为起草说明(preamble)。
起草说明为何这么长呢?在美国的法规系统中,它扮演什么角色呢?
首先,规章是法律的细化和延伸,是强制性内容。其行文重在严谨,而非宜读。规章有规范、警示和惩罚的作用。规章的读者,通常是非法律专业的企业人员,若是不能吃透法律上严谨的文字,就达不到规范和警示的作用了。起草说明,就是用普通文字来详述规章制定或修订的来龙去脉和斟酌思考的。这是美国法律系统的传统。例如,最高法庭断案的结论可能只有短短几行专业、严谨但难读的文字,但赞同派和反对派的法官们会分别写出几十甚至上百页的意见书,详述各自的斟酌思考。这些意见书,都是用普通易懂语言写的,旁引博证,掰开来、揉过去地讲,目的是争取公众的理解和支持。
也许有人会问,指南不就是用来解释规章的吗,为何还需要起草说明呢?
这可以从如下两个角度来理解。一,可以将起草说明看成是在规章制定或修订之初的指南,或01版指南。二,指南是FDA从自身角度对规章的释意,而起草说明往往包含大量的对企业提出的具体问题的具体回复,针对性和启发性强。例如,本文讨论的这两项拟修订规章的核心,是如何判断标签外用药是否是企业的意向。现在来看看起草说明是如何通过具体示例来说明FDA是如何判断的。
是不是药或器械,不取决于名称
产品是否应按照药品或器械来监管,不看厂家是否说它是药品或器械,甚至不看标签上是否这样说,而看产品的意向用途(intended use)。只要意向用途是治病防病,就应按照药品或器械来监管。对意向用途判断的证据,来自包括产品营销的所有相关材料和言行。
证据的相关性,而不是决定性
规章中所说的意向性产品用途的证据,任何一项单独看,都只表示可能的相关性(may be relevant),而不构成决定性证据(not determinative)。
间接的意向性证据的举例
- 产品名称上的暗示,例如,产品名称Chronix暗示慢性(chronical)病,产品名称Shroomz暗示借助毒mushroom(蘑菇)中的药性,产品名称e-Cialis暗示壮阳药(Cialis,希爱力-他达拉非,是礼来公司治疗男性勃起功能障碍的药)。
- 企业对已上市药品,做了增加适应症的新临床试验,事后写给参加临床试验的医务人员总结报告,不是客观、完整、正反面都说的,而且包含了对新适应症的安全和有效性做出结论性的言语。
企业做法的一贯性
判断企业对批准产品的非批准用途的意向性,要看企业的做法是否是repeated(多次的),proactive(主动的)和detailed(详尽的)。
上面讨论的是起草说明对企业的作用。那它对FDA自身有什么作用呢?
起草说明中篇幅较大的另外两项分别是背景介绍(30%)和经济影响评估(18%)。背景介绍讲的是规章制定和修订过程中发生的主要事件、缘由和处理。这种来龙去脉的详细介绍,不仅有助于对规章的系统和深入的了解,而且展示了FDA政策的连贯性。制定规章时,开展经济影响评估,尤其是对小企业的经济影响评估,体现了规章的合理性。所有这些,都有助于加强和巩固公众对FDA的信任感。药品法只给了FDA制定、执行和解释规章的法律权利。但FDA的做法是否服众,是要靠自己挣的。
对FDA,写好起草说明,是制定规章时必不可少的一步。对企业和公众,研读起草说明,是系统和深入地了解规章的良习。再举一例,FDA的药品GMP,1978年在联邦公报上发布时,长达76页纸。前63页纸是起草说明,包含了对在制定规章过程中收到的518条意见的详细回复,解释了FDA是如何看待这些回复的,哪些地方同意,哪些地方不同意,理由是什么。这样制定出的规章,容易科学合理,容易符合产业发展规律,容易为产业接受。也许,这就是40多年来,FDA的GMP一直未有大修订的原因?
对阅读起草说明有兴趣的读者,可登录识林阅读本文所讨论的这两篇起草说明。
作者:榆木疙瘩
识林®版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
解读法规指南 适用岗位: - QA(质量保证部门)
- 注册部门
- 市场准入部门
- 研发部门
- 临床研究部门
工作建议: - QA:确保所有产品标签符合21 CFR 201的规定,监控标签的变更和审核流程。
- 注册部门:在提交药品注册资料时,包括标签设计,确保其符合FDA的标签要求。
- 市场准入部门:在市场推广材料中使用标签信息时,确保信息的准确性和合规性。
- 研发部门:在药品开发阶段考虑标签要求,以确保最终产品的标签合规。
- 临床研究部门:确保临床试验中的药品标签正确反映了试验要求。
适用范围:
本文适用于在美国市场的所有处方药和非处方药(OTC)药品,包括化学药品、生物制品、疫苗等。适用于所有在美国进行药品研发、生产、注册和销售的企业,包括Biotech、大型药企、跨国药企、CRO和CDMO等。 要点总结: - 标签信息要求:强调了药品标签上必须包含的信息,如活性成分、使用说明、警告、剂量形式和强度等。
- 净含量声明:规定了药品标签上净含量的表达方式,包括重量、度量或计数,并要求准确反映包装内药品或设备的数量。
- 警告和注意事项:要求药品标签上必须有清晰的警告和使用上的注意事项,以确保患者安全使用。
- 特殊人群用药:对孕妇、哺乳期妇女、儿科和老年用药进行了特别规定,要求标签上提供相应的用药信息和警告。
- 药品相互作用:要求标签上必须包含可能的药品相互作用信息,以帮助医疗专业人员和患者了解潜在的风险。
以上仅为部分要点,请阅读原文,深入理解监管要求。 法规指南解读适用岗位- QA:必读,确保产品标签符合FDA规定。
- 注册:必读,了解标签要求以支持产品注册。
- 市场:必读,确保市场推广材料符合标签规定。
工作建议- QA:检查所有产品标签,确保符合FDA CFR 801的要求。
- 注册:在产品注册文件中包含符合CFR 801的标签信息。
- 市场:确保所有市场材料不包含误导性信息,与产品标签一致。
适用范围本文适用于美国FDA监管的医疗器械的标签要求,涉及所有类型的医疗器械,包括创新医疗器械和仿制医疗器械。 要点总结- 标签信息完整性:强调了医疗器械标签必须包含完整的产品信息,包括名称、用途、预期用户等。
- 风险信息明确:要求标签上明确标注产品的风险信息,以便用户做出明智决策。
- 使用说明详细:标签应提供详细的使用说明,确保用户正确使用产品。
- 警告与预防措施:标签上必须包含必要的警告和预防措施,以减少产品使用过程中的风险。
- 监管要求遵守:强调企业必须遵守FDA的监管要求,确保产品标签的合规性。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |