首页
>
资讯
>
【周末杂谈】OAI、透明公开监管与产业发展
出自识林
2020-01-12
上月11日,在美国食品药品法研究所在华盛顿举行的一次会议上,FDA药品审评和研究中心合规办公室(Office of Compliance,OC)主任D. Ashley高兴地报告,2019财年(9月30日截至),在GMP检查后90天(本文引用的都是自然天,不是工作日)内,OC完成了86%的检查结果分类,比2018财年增加了4个百分点,得到了业内普遍的赞赏。
通常GMP检查完成后,检查员和检查派出机构 - 监管事务办公室(Office of Regulatory Affairs,ORA),会对企业的GMP合规状态,给出分类建议,即NAI(No Action Indicated, FDA认可企业的GMP合规),VAI(Voluntary Action Indicated, FDA认为GMP合规虽有不足,但企业自行改进便可),或OAI(Official Action Indicated, FDA认为GMP合规严重不足,需要监管制裁,包括发警告信、进口禁令、甚至扣押上市产品等)。OC依据ORA的建议及自身对企业的了解,包括企业的483回复信和相关资料和数据,最终决定是NAI,VAI还是OAI。
零483固然好,但不被分类成OAI才是关键。若483中包含严重的违规项,例如:数据可靠性 问题,OAI分类的可能性增大。一旦分类成OAI,下一步就可能会收到警告信、进口禁令 或更严重的制裁,从而严重影响企业的运行和运作。因此,企业通常希望FDA尽快决定最终分类并通知企业,从而早做应对准备,例如,找其它委托生产 商。
鉴于合规分类的重要性,经行业与FDA协商,涵盖2017-2022年的仿制药使用者付费法案II期(Generic Drug User Fee Amendments II,GDUFA II) 要求FDA在检查后90天内确定分类,并在此后90天内做出监管制裁。后来,FDA将这两项要求的适用范围推广到包含创新药。
从透明和公开的角度看,Ashley主任的乐观不无道理。透明是让行业监督,公开是让行业参与。行业既是监管的对象也是监管服务的顾客。顾客的满意度,是监管成效的量度。顾客的监督是降低监管政策制定和执行中出差错风险的低成本和高效率的手段。FDA的各种使用者付费法案是践行透明和公开的良好案例。法案中将行业对FDA期望和FDA的承诺,落实到具体量化指标,公之于众,并要求FDA每年向国会和行业汇报,逐项说明完成承诺的情况。
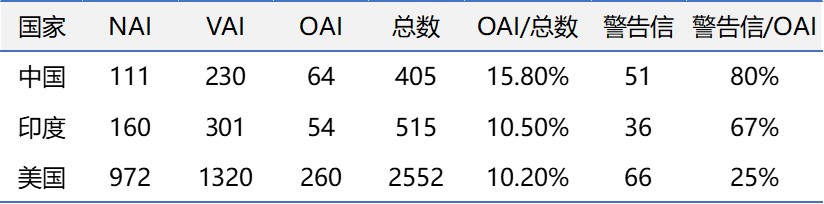
指标量化和公开后,就可以问深入一些的问题了。例如,Ashley主任的报告中间接提到2018财年未按时完成分类的比例是18%(比2019财年的14%高4个百分点)。下表是2016-2018自然年的FDA分类数据。虽然两者时间区间不完全吻合,但取三年的平均值提高数据的信噪比。FDA对中国企业的GMP检查,OAI分类占总数的15.8%。但愿中国企业没这么倒霉,摊上了所有延迟的OAI分类。两个数,15.8%与18%接近,恐怕是巧合,只有FDA准确知道。
表中最后一列显示的,恐怕就不一定是巧合了。美国企业被OAI分类后,只有25%吃警告信。印度企业则高达67%,中国企业更是高到80%,所谓十有八九。这种显著差异,可以有多种解释。其中一种,是中国企业化险为夷的能力比美国和印度企业差,反应在自我认知程度、科学和法规的认知水平、利用外部资源能力、及理性客观交流沟通能力等方面的差距。这方面,前任OC主任Tom Cosgrove曾具体讨论过,周末杂谈今后将专题报道。
作者:榆木疙瘩® 版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。