首页
>
资讯
>
分析报告显示 FDA 对海外药品生产监管力度仍远低于疫前水平
出自识林
分析报告显示 FDA 对海外药品生产监管力度仍远低于疫前水平
2023-12-06
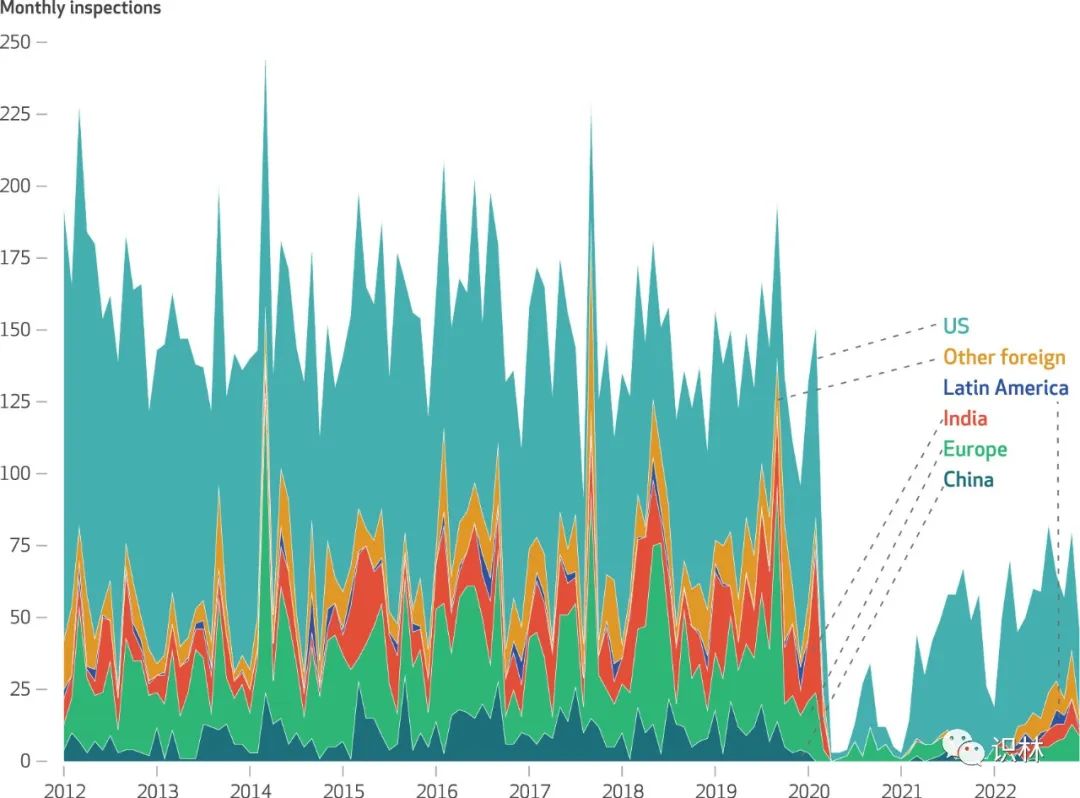
根据 12 月 4 日发表在《卫生事务》(Health Affairs)上的一篇题为“FDA 全球药品检查:对生产企业的监督仍远低于新冠(COVID-19)之前的水平”(FDA Global Drug Inspections: Surveillance Of Manufacturing Establishments Remains Well Below Pre-COVID-19 Levels)的分析文章,新冠疫情之后,尽管生产活动并未下降,但美国 FDA 所检查的本土和海外制药场地大幅减少。与此同时,FDA 的预算和人员配置保持稳定,因而分配给每次检查的资源激增,并且更多工厂被指严重违规。
具体而言,2019 年至 2022 年间,美国以外的药品生产工厂的检查量下降了 79%,本土工厂的检查量下降了 35%(图1)。同时,在美国本土、欧洲、印度和中国分配检查的平均检查人员数量和每次检查的持续时间有所增加,在印度和中国的检查去年略有回落。
另外,疫情的爆发也标志着 FDA 检查员确定需要采取补救措施的工厂数量猛增,2021 年这一数据占所有本土检查总数的 27%,而此前一年占比仅为 12%。不过 2022 年这一占比回落至16%。对于海外工厂,过去两年中进行的所有检查中约有 17% 收到警告,而 2020 年这一比例仅为 5%。
这些分析结果凸显了 FDA 在试图摆脱疫情影响并恢复惯常的检查方法时所面临的困难,同时也有新的紧迫感。尽管如此,研究作者认为,FDA 本质上未能满足加强监管的需求,因此需要替代方法来确保药物安全。
该研究的合著者之一,杜克大学专门研究药物创新、生产和定价的 David Ridley 表示,“这是一个问题,并且会随着时间的推移而恶化。检查之间的间隔越长,出现未解决问题的可能性就越大。”
这一分析研究再次强调了人们长期以来对 FDA 监控美国药品供应链关键组成部分的能力的疑虑。这已不是新话题。去年初,美国政府问责办公室(GAO)指出了自 2009 年以来出现的问题 ,并表示 FDA 面临“独特的挑战”,尤其是在招聘和留住检查员方面。与此同时,由于对 FDA 监管不确定性的担心,美国国防部和凯撒医疗机构最近开始与私人实验室合作检测药品的安全性 。
今年七月份,众议院能源和商业委员会要求 FDA 提供有关中国和印度制药工厂检查的详细信息 。委员会指出,这些国家的一些公司“屡次违反 FDA 安全性法规。”
政策建议
研究指出,随着全球疫情结束,FDA 必须解决积压的检查问题并考虑未来的新方法。文章提出了五项建议,包括:
增加检查员队伍。 2016 年《21 世纪医药法案》 赋予 FDA 一定的权力来招聘难以填补的职位。如果还不够,国会应给予 FDA 更大的招聘灵活性。更多的工作人员可以进行更多的检查、缩短检查之间的间隔以及检查员在不同场地之间的轮换,这对于消除因同一检查员重复检查同一场地而产生的自满情绪至关重要。
优先考虑海外检查。 自 2020 年 3 月以来,FDA 推迟了非必要的海外检查,造成大量积压。优先进行海外检查将缩小因疫情而重新出现的美国本土和海外场地的检查差距。
加强国际合作。 FDA 应继续使用互认协议,承认美国以外国家和工厂所在国家监管机构的检查报告和产品抽样计划。此外,FDA 应继续在进行现场检查之前或不进行现场检查时要求提供记录,并使用视频通话和屏幕共享等技术来审查无法进入的工厂的数据。但这些努力应该补充而不是替代未来的海外现场检查。
分散药品质量保证 负担。 例如,凯撒医疗和国防部目前正在与实验室合作检测药物并根据质量选择生产商。其他中介机构,例如批发商和零售商,也应该效仿。尽管第三方无法确定特定制造企业的质量,但他们可以深入了解特定公司的平均质量。被认定为销售高质量产品的公司的举措可以为这些公司带来更大的市场份额和更高的价格。正如时任 FDA 药物审评和研究中心主任的 Janet Woodcock 曾说过的,“检查不是万能药。……让企业对质量本身负责很重要。”
提高患者对药品质量的意识。 应让患者了解药品质量问题。如果患者了解仿制药 制造商之间的平均质量差异,那么他们就可以做出基于质量的购买决定。Ridley 表示,“我们目标不是让每个消费者都积极寻找优质药品,而是让很大一部分消费者这样做,从而影响市场趋势。理想情况下,零售商可以通过专门储存高质量的仿制药、简化消费者的流程并推动更广泛的市场变革以优先考虑药品质量来使自己脱颖而出。”
作者:识林-椒
识林® 版权所有,未经许可不得转载
岗位必读建议:
研发(R&D):了解加速药品开发流程的新规定,确保研发项目符合新法规要求。 注册(Regulatory Affairs):掌握法规对药品注册流程的影响,优化注册策略。 临床(Clinical):关注临床试验设计的新指导原则,确保试验合规性。 文件适用范围:
文件要点总结:
加速药品开发流程 :强调了加速药品从发现到上市的整个流程,以促进21世纪医疗创新。临床试验现代化 :提出了对临床试验设计的现代化要求,以提高试验效率和患者参与度。个性化医疗推进 :鼓励发展个性化医疗方法,包括精准医疗和基因疗法。数据共享与隐私保护 :规定了数据共享机制,同时强调了患者数据的隐私保护。监管框架更新 :明确了对FDA监管框架的更新,以适应新兴医疗技术和产品。以上仅为部分要点,请阅读原文,深入理解监管要求。