首页
>
资讯
>
可见颗粒物污染的生命周期解决方案
出自识林
2020-01-13
可见的颗粒 物污染是导致注射剂批次被拒绝或召回的主要原因之一,在2009年至2019年间,发生在美国的注射剂召回事件中,存在异物颗粒问题是仅次于缺乏无菌 性保证的原因。美国注射剂协会(Parenteral Drug Association, PDA)2015年对注射剂生产商的调查显示,上年度:
43%的受访者由于颗粒物而对1-5个产品批次进行了重新检查, 18%的受访者重新检查的批次超过了10个。
50%的受访者发生过产品批次拒绝。32%的受访者拒绝过1-3个批次,其余则超过了3个批次。
26%的受访者表示收到过来自监管机构有关颗粒物控制的观察结果。
9%的受访者表示在过去一年中由于成品中的颗粒物问题召回 了一个或多个批次。
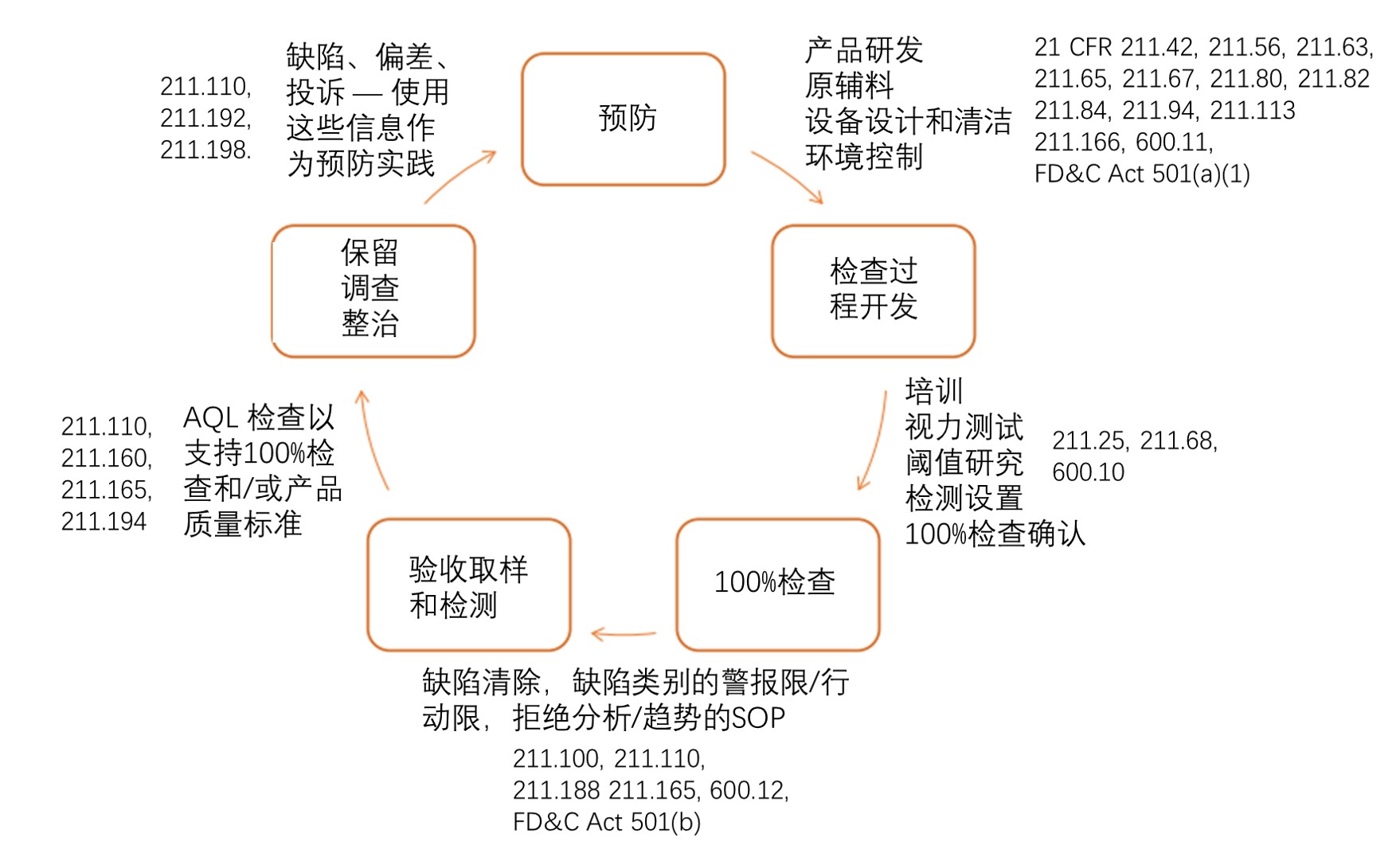
颗粒污染的结果会造成时间、精力、材料、资源、金钱、产品的浪费,以及由于产品不可放行或召回而引起药物短缺 。在PDA Journal 在2019年接收的文章中[1],总结了一种控制可见颗粒污染的生命周期 方法(见下图)。预防可见颗粒污染,要从产品开发开始,首先通过评估产品属性,容器密闭件系统特性和稳定性限制因素;需要控制生产环境、容器/密闭件系统处理过程、产品组件和生产设备;还需要充分的检验方法开发、培训 和资质确认以验证和执行目检;最后评估目检获得的信息,将其用于防患于未然。该方法与FDA CGMP要求一致,是检查、审计时证明监管合规的有效工具。这种整体的可见颗粒控制方法应与质量风险管理计划结合使用,考虑可见颗粒污染的来源,以加强检查方法的开发,确保每批成品从根本上不含可见颗粒。
预防:
研发时应当表征产品中可能存在的固有可见颗粒,例如脂质体 、悬浮液和蛋白聚集体,应有相应的人工或自动目检技术以及提供训练方法,以区分固有颗粒物与异物(例如,“麦粒状”固有蛋白颗粒物与外源纤维的区别)。研发还应基于药物特性(例如pH值 ,离子强度,缓冲液系统和灭菌 方法)以及储存条件和使用方式等来预测、表述固有颗粒(来自产品,工艺或容器系统的微粒)形成的可能性,例如玻璃薄片或玻璃容器与药品发生反应而产生的沉淀物 (21 CFR (§)211.94 (a) , §600.11(h), §211.166 )。
生产工艺应经过良好的设计,将产生可见颗粒污染的风险降至最低。生产设施、设备等必须适用于防止可见颗粒的污染,并且可进行充分的维护、清洁和操作(§211.42 ,§211.56 ,§211.63 ,§211.65 ,§211.67,§211.113,§600.11)。例如,通过使用HEPA过滤器、清洁、维护墙壁和天花板、洁净服良好实践以及操作员培训来控制无菌操作区中的空气传播污染物,防止携带微生物的颗粒进入产品容器;通过适当的设施设计和设备维护,将叶轮磨削、灌装针头撞击或玻璃破裂等事件产生的金属或玻璃颗粒降至最低;制定接收 、鉴别 、贮存 、处理、检验 、隔离 、取样 、检测、防止污染以及批准/拒绝原辅料 和容器密闭件组件的书面规程(例如玻璃瓶包装中有破损的小瓶或者对生产环境有其他影响 (§211.80, §211.82))容易被污染的种类必须进行采样 ,根据这些污染物的既定标准进行检测。
检查过程开发:
注射剂等的目检需要考虑产品特性、容器类型、检查方法以及该方法要检查的缺陷 类型,对于可见颗粒的检查,无论是使用人工、半自动还是全自动系统,都可以根据缺陷类别(如裂缝、压塞缺陷、粉饼缺陷)整合为整体目检过程。还要进行阈值研究确定代表性产品和容器系统中不同类别颗粒(如玻璃、纤维、橡胶等)的检出限。有些产品的检查会有些困难,例如冻干产品、带有外包装的软袋、使用半透明或不透明容器的产品,单靠100%目检是不行的,还需要做破坏性检测才能满足药典要求。
半自动或全自动系统的检查可以增大吞吐量,减少人工劳力,并且可以应用人工智能/深度学习改进检查过程。然而,每种缺陷对应的检测设备应该相匹配,且按照相应的SOP 执行预期功能,并保留常规设备校验 和性能确认 的书面记录 (§ 211.68)。
参与生产和检查的人员都应经过培训,才能执行其指定的职能,除了预防颗粒污染方面的培训还需要眼科检查、目检再确认 等对最终产品的检查,而且这种培训是持续的(§211.25,§600.10)。
100%检查:
USP第<1>章注射剂和植入式药品(肠胃外药物)产品质量检查指出:
1. 应对所有肠胃外制剂的每个最终容器进行检查,以确保其内容物中是否存在可观察到的异物和颗粒物(以下称为可见颗粒)。
USP第<1>章提到100%检查, USP第<790>章指出通过100%检查过程在AQL检测之前剔除缺陷单元,因此100%检查中涉及人员必须经过资质确认,涉及到的设备也必须经过确认以保证符合药典标准(FD&C法案 501(b), §211.165)。如211.100条款所述,质量部门应实施为保证产品质量而制定的生产和工艺控制书面规程。包括在生产之前和期间防止颗粒污染的规程,以及检查成品的规程。
根据历史数据确定可见颗粒污染率的中间过程质量标准 ,并且应与最终产品质量标准相协调一致(§211.110)。将可见颗粒的中间过程结果和实验室检测结果记录在批记录中(§211.188, §600.12)。
生产商需要将可见颗粒污染和其他可见缺陷分为严重,主要和次要缺陷,并建立可接受限度。通常认为按照产品标签中的指示使用可能对患者造成严重不良事件 的那些缺陷为严重缺陷,如有裂纹、泄露或出现可见颗粒等表明该产品的无菌性无法保证;某些与CGMP 违规无关的固有颗粒,通常引发不良事件的可能性很小,并不预示无菌保障的失败,仅仅导致产品单元报废,则属于主要缺陷,但还应考虑诸如目标患者人群、给药途径 和给药频率等因素;容器瑕疵或其他外观缺陷不会影响患者安全,但可能会导致单元报废,属于轻微缺陷。利用这些缺陷的趋势,及早发现工艺控制问题来确保批次质量和均一性。
验收取样和检测:
验收取样和检测是在产品批次放行前,确认预防措施和100%检查足以控制可见颗粒污染的程序。USP第<790>章注射剂可见颗粒指明在批次放行时进行AQL检测,但不作为批次放行的条件。如果已批准的产品质量标准参引了USP第<1>章和第<790>章作为可见颗粒检查的方法和/或可接受标准,那么每个批次的药品都要经过实验室检测确保符合质量标准(§211.165),这包括取样和检测书面规程以及测试的样品数量。生产商必须验证和记录在实际使用条件下检测方法的准确性、敏感度、特异性和重现性(§211.194(a)(2)),拒绝不符合质量标准 的批次。在有适当的科学依据、规程和质量部门批准的情况下,可以对未通过初次AQL检测的批次进行重新检测。尽管USP第<790>章提供了最低的AQL(生产商风险)接受标准,但在决定接受或拒绝每批产品时也必须考虑UQL(患者风险)。为确保批间均一性,需建立书面规程,对每个批次样品实施中间过程控制、检测,以监测输出和验证工艺性能(§211.110)。从每个批次中进行取样是很重要的,因为这些样品被用以确认可见颗粒污染的预防情况以及100%检查的适用性。
保留、调查和整治:
对在100%检查或AQL检测过程中被剔除的含有可见颗粒的单元,按照颗粒污染的类型、来源和严重性进行趋势分析。在批次放行和发运前,要检查所有与可见微粒相关的生产和控制记录确保符合已批准的书面规程。即便是发运后,发现任何无合理解释的批次或组件(胶塞、容器、原料等)的缺陷或失败也都要进行调查,调查可以延伸到这类缺陷或失败的其他受影响批次,同时做好调查记录和总结(§211.192)。建立处理与可见颗粒污染有关的口头和书面投诉的规程,并对所有投诉进行书面记录(§211.198),规程还需要包括对投诉 、召回 、退货样品的回顾,以及当有代表性数量的产品批次足以确定已建立的质量标准、生产或工艺控制发生了变化需要进行变更的规程。通过100%检查、AQL检查、客户投诉和稳定性样品获得的信息,用以确定可见颗粒污染的来源和改进生产工艺。对这些被拒绝单元中的颗粒进行分析,可以得到在现有生产环境下,出现的可见颗粒缺陷类型和频率的有价值的信息。此类信息可用来整治可见颗粒问题、加强人工或自动检测。
相关警告信:
如果未按照监管要求处理生命周期中可见颗粒事宜,违反相关条款,就会有触发警告信的风险。在识林警告信数据库 中搜索“可见颗粒/微粒/异物”,通过具体的案例学习该如何做才能从根本上保证注射剂中不含颗粒物。
相关阅读
2014年,美国药典(USP)发布了第<790>章 注射剂中的可见颗粒,提供了一种检测方法(基于欧洲药典2.9.20章)和被认定为从根本上没有可见颗粒药品批次的最低接受限度。还提供了可见颗粒检查的参考指南,可用于内部检查流程或将其包含在成品质量标准中。
2017年,USP发布了<1790>注射剂的外观检查,其中提供了有关注射药品的整体外观检查流程的更多信息。
2019年,USP42-NF37 第<1>章 注射剂和植入药品(非肠道用药)– 产品质量检测。
参考资料
适用岗位必读指南:
QA:负责确保所有操作符合cGMP要求,包括生产、质量控制、设备维护等。 生产:必须遵守书面程序,确保产品质量。 质量控制(QC):负责样品的测试和批准或拒绝,以及稳定性测试。 设备维护:确保设备清洁、维护和校准符合规定。 仓储与分销:遵守药品存储和分发的书面程序。 文件适用范围:
文件要点总结:
质量控制单元的责任: 必须有一个质量控制单元,负责批准或拒绝所有组件、药品容器、包装材料、标签和药品,并审查生产记录以确保没有错误发生或错误已得到全面调查。
人员资质与责任: 参与药品生产、加工、包装或储存的人员必须具备相应的教育、培训和经验,并遵守良好的卫生习惯。
设备设计、清洁与维护: 设备应适当设计,便于操作、清洁和维护,并按规定进行定期清洁和维护。
组件和药品容器的控制: 必须有书面程序详细描述组件、药品容器和闭合件的接收、识别、存储、取样、测试和批准或拒绝。
生产和过程控制: 必须有书面程序确保药品具有其声称或代表的身份、强度、质量和纯度,包括偏差处理和产量计算。
包装与标签控制: 必须有书面程序确保正确的标签和包装材料用于药品,包括防篡改包装要求。
仓储与分销程序: 必须有书面程序描述药品的存储和分发,确保药品质量。
实验室控制: 必须建立科学合理的规格、标准、抽样计划和测试程序,以确保药品及其组件符合适当的身份、强度、质量和纯度标准。
记录与报告: 所有与生产、控制或分发相关的记录必须保存至少一年,或在特定情况下保存更长时间,并随时可供授权检查。
退回和报废药品的处理: 退回的药品必须被识别并保留,除非证明其符合适当的安全、身份、强度、质量和纯度标准,否则应销毁。
以上仅为部分要点,请阅读原文,深入理解监管要求。