首页
>
资讯
>
国内药政每周导读:临床审评标准,新增三个分中心,化药新药晶型,Q3E 中文版,医保局真实世界价值研究
出自识林
国内药政每周导读:临床审评标准,新增三个分中心,化药新药晶型,Q3E 中文版,医保局真实世界价值研究
2025-09-29
【早期开发与临床】
9.28,【CDE】关于公开征求《药物临床试验申请临床评价技术指导原则(征求意见稿)》意见的通知
征求意见截至10月28日。
本指导原则 起草过程中,详细梳理了药物临床试验申请时临床评价的关键技术考虑,并借鉴参考了国内外监管机构、ICH 等发布的相关指导原则、已形成的《化学药品及治疗用生物制品临床试验申请临床专业审评报告撰写规范》等内部资料。
本指导原则分为四个章节,包括概述、总体考虑、临床评价的主要内容和参考文献。
“临床评价的主要内容”是本指导原则的核心,系统阐述药物临床试验申请时临床评价的关键技术考虑,并对临床需求评估、前期研究数据评价、研发计划和临床试验方案 的评价、临床试验规范及相关风险控制、临床试验 过程中安全性评估等评价关注点进行了详细描述。
本指导原则中,对于“(四)临床试验方案评价”主要包括 I 期临床试验方案及 II/III 期临床试验方案的评价考量。在对 II/III 期临床试验方案的评价中,强调了在启动 III 期临床试验前,应评价研究药物所有已开展的临床试验数据,并应关注支持 III 期临床试验的前期证据是否充分,该部分已包括对 II 期临床试验方案及结果评价的要求,同时考虑 II 期和 III 期临床试验方案评价相近,故予以合并未再单独列出 II 期临床试验方案评价考虑。
【CMC与仿制药】
9.23,【CDE】关于公开征求《化学药品创新药晶型研究技术指导原则(征求意见稿)》意见的通知
征求意见截至10月23日。
本指导原则 主要根据创新药的特点和晶型 研究的现状,重点阐述了晶型的选择与表征、原料药 的晶型控制、制剂中的药物晶型控制等药学研究的关注点及一般原则,为化学药品创新药晶型研究的研发提供技术指导。
本指导原则主要分为六个部分,分别为概述、总体考虑、晶型的选择与表征、原料药的晶型控制、制剂中的药物晶型控制和参考文献。
9.26,【CDE】关于公开征求 ICH《Q3E:可提取物与浸出物指导原则》指导原则草案及其支持性文件意见的通知
ICH Q3E征求意见稿中文版 截至12月15日。
ICH征求意见稿 发布于8月7日,可见专题文章《ICH 新指南草案 Q3E:可提取物与浸出物的控制框架和具体各论》 。
【注册与变更】
9.24,【CDE】常见一般性技术问题解答 - 新增1个问答
Q:“药品注册批件及其附件”中存在文字性错误,可通过什么途径沟通解决?
A:为保证勘误处理时效,自2025年9月25日起,申请人 可通过国家药品监督管理局政务服务平台,提交“药品注册批件及其附件”的勘误申请,其中涉及药审中心职责的,药审中心在规定时间内核实处理,原则上不再接收纸质版勘误申请。
【经营准入】
9.23,【国家医疗保障局】关于开展真实世界医保综合价值评价试点工作的通知
国家医疗保障局发布通知,旨在推进真实世界医保综合价值评价试点工作,以科学化医保治理体系,助推医疗医药高质量发展。
试点工作关注药品审评审批或药品目录谈判中未充分回答的医保关切问题,以及药物在真实世界 应用后的实际临床获益和风险。试点任务分为启动、实践、应用三个阶段,目标是到2027年底形成一套基于真实世界研究的医保综合价值评价体系,并在全国推广。
对于药品,其应用场景包括:
【监管综合】
9.24,【NMPA】药品和医疗器械审评检查京津冀分中心、华中分中心、西南分中心挂牌成立
9月21日至24日,国家药监局分别在北京市、湖北省武汉市、重庆市举行药品和医疗器械审评检查京津冀分中心、华中分中心、西南分中心挂牌仪式。国家药监局党组书记、局长李利一行参加挂牌仪式并调研当地医药产业发展、分中心筹建等情况。北京市委副书记、市长殷勇,湖北省委副书记、省长李殿勋,重庆市委副书记、市长胡衡华出席挂牌仪式。国家药监局党组成员、副局长黄果在挂牌仪式上致辞,局党组成员、副局长雷平主持。
【新药批准和报产】
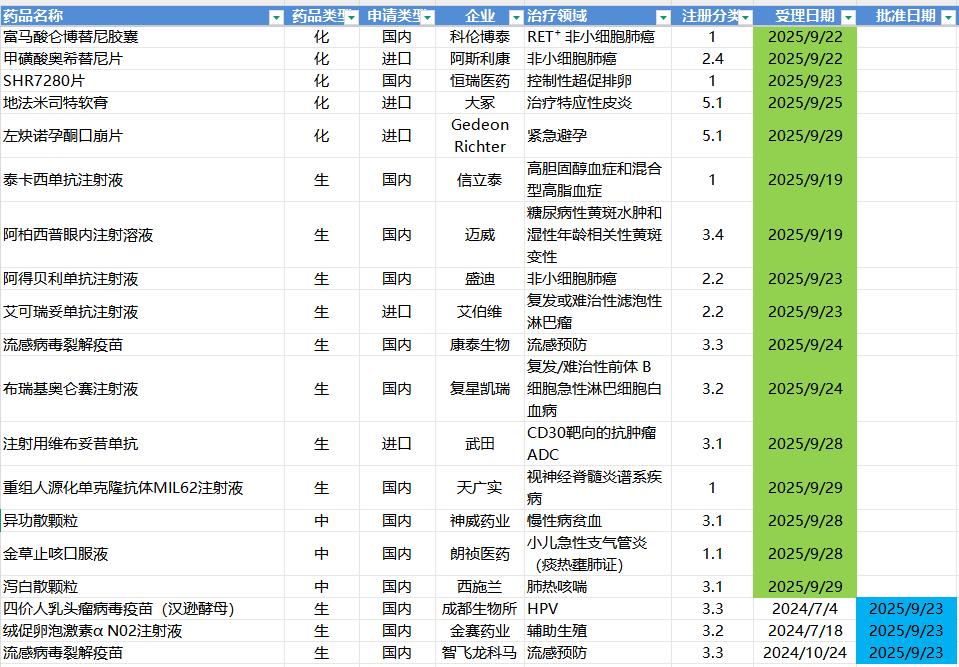
9.22-9.28,NMPA发布3个新药批准,CDE受理16个新药上市申请
注:仅列出新药(包括改良型新药 )的上市申请和批准上市信息。在“以临床价值为导向”的背景下,申请上市以及获批的品种,其适应症 、临床研究 策略、注册路径,都值得业界关注和分析。
识林® 版权所有,未经许可不得转载
适用岗位及工作建议:
QA(质量保证):必读。需根据文件更新质量控制流程,确保生产组件、包装和给药装置符合风险评估和控制要求。 注册(注册事务):必读。负责在药品注册文件中包含可提取物/浸出物研究的合理性论证、相关研究报告和风险控制策略。 研发(研发部门):必读。在新药制剂开发过程中,需遵循风险管理原则,评估和控制浸出物风险,保障患者安全和产品质量。 生产(生产部门):必读。在生产过程中,应实施风险降低措施,如更换组件/供应商、预清洗等,并建立适当的控制策略。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
研发(R&D) :必读。在药物研发过程中,应重视晶型研究,选择优势晶型,并制定晶型控制策略。质量控制(QC) :必读。需根据ICH Q6A评估晶型控制,并在质量标准中制定晶型鉴别和检查项。生产(Production) :必读。应根据目标晶型的理化特性与稳定性,制定合理的工艺控制策略,保证原料药的生产工艺可稳健获得目标晶型。适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
【文件概要】
【适用范围】
【影响评估】
【实施建议】
注册 :必读。需对照文件调整申报策略,如多肽类制剂按境内外上市情况选择分类,境外变更文件豁免需符合指南条件。 QA :必读。关注非无菌制剂微生物控制标准更新,确保生产工艺与注册标准一致。 研发 :必读。罕见病药物研发需评估是否符合临床试验减免条件,共晶制剂需明确活性成分分类依据。 临床 :必读。临床试验期间剂型变更需重新申请,早期试验适应症填写需备注多队列研究细节。 生产 :必读。批生产记录需包含关键批次,电子申报资料需严格遵循光盘刻录及签章规范。 以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及建议:
临床(Clin) :必读。应根据指导原则评估临床试验方案,确保科学性和安全性。注册(Reg) :必读。需理解并应用指导原则中的要求,以准备和提交临床试验申请。研发(R&D) :必读。在药物研发早期阶段,应依据指导原则考虑临床需求和前期研究数据。适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
QA(质量保证):负责确保文件中关于leachables和extractables的控制措施得到实施,并监督相关质量风险管理流程。 RA(注册事务):在药品注册过程中,需要参考本文件准备和提交leachables和extractables的相关研究资料。 R&D(研发):在新药研发阶段,需依据本文件评估和控制leachables,确保药品的安全性和质量。 CMC(化学、制造和控制):负责执行leachables和extractables的具体研究工作,并确保数据的准确性和完整性。 工作建议:
QA:制定和更新内部SOP,以符合指南要求,并对相关流程进行审计,确保合规性。 RA:在注册文件中包含leachables和extractables的风险评估和控制策略,与监管机构沟通以获得批准。 R&D:在药物开发早期阶段进行leachables的风险评估,并选择合适的包装和制造组件以最小化风险。 CMC:进行leachables和extractables的实验研究,提供准确的数据支持药品安全性评估。 适用范围:
要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。 QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:本文件适用于医保政策制定人员、药品注册人员、市场准入人员、研发人员和临床研究人员。对于医保政策制定人员,建议深入理解文件中关于医保综合价值评价的总体原则和试点任务,以便制定符合国家医保局要求的医保政策。药品注册人员应关注真实世界数据在药品审评审批中的应用,以优化药品注册流程。市场准入人员需了解评价体系对药品市场准入的影响,以制定有效的市场策略。研发人员应考虑如何在研发阶段收集和利用真实世界数据,以支持药品的综合价值评价。临床研究人员应关注真实世界研究的设计和实施,以提供高质量的临床数据。
适用范围:本文适用于化学药、生物制品等药品类型,涉及创新药、仿制药、生物类似药等注册分类,发布机构为中国国家医疗保障局,适用于大型药企、跨国药企、Biotech等企业类别。
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。