首页
>
资讯
>
国内药政每周导读:NMPA药品年报制度落地,PV检查正式发文,双抗肿瘤药临床指南,药典2022年标准提高课题
出自识林
国内药政每周导读:NMPA药品年报制度落地,PV检查正式发文,双抗肿瘤药临床指南,药典2022年标准提高课题
2022-04-18
【注册、审评、审批】
4.14,CDE更新13个品种的23篇上市药品信息(说明书与审评报告)
【临床研究】
4.11,CDE征求《双特异性抗体类抗肿瘤药物临床研发技术指导原则》意见
双特异抗体 (bispecific antibody,BsAb)目前是抗肿瘤新药的研发热点之一,尤其是在肿瘤治疗领域,BsAb的药物研发 持续增长。
单抗 类抗肿瘤药物的临床研发思路与技术要求已较为成熟,但对于针对多个抗原的BsAb,由于结构与功能存在特殊性,因此在临床研发中,有其特殊考虑要点。本指导原则旨在为BsAb类抗肿瘤药物的临床研发中,需要特殊关注的问题提出建议。
BsAb的临床研发首先应遵循一般药物研发原则及规律,但同时BsAb作为一类具有双功能的“单药”,既不同于有关的单抗,也不同于单抗的联合用药。对于针对多种抗原表位的BsAb,由于结构与功能存在特殊性,因此在临床研发中,有其特殊考虑要点。
另一方面,开发BsAb的目的,是通过解决单抗不能解决的治疗问题,为患者带来单抗治疗所不具备的临床获益;BsAb的研发遵循着药物结构决定作用机制,从而决定临床获益风险特征的规律,因此BsAb的开发,应该体现以解决单抗不能解决的问题为主要目标,以临床需求为导向的抗体设计和开发思路。
【CMC药学研究】
4.11,药典委发布《2022年度国家药品标准提高项目课题目录》
按照国家药典委员会《药品标准制修订研究课题管理办法》 (2018年11月,药典委发,以下简称“管理办法”),经公开征集课题建议及承担单位、组织专业委员会及专家组审议、网上公示、药典委审核等程序,确定了2022年度国家药品标准提高任务。
企业需确认是否有与自身业务相关的标准提升,提前做好相应准备,可关注其2个附件:
1、2022年度国家药品标准提高项目课题目录(品种)
包括83个中药、化药、药用辅料 ,诸如:
银杏叶提取物、滴丸中双黄酮成分限量检查方法;
吸入用盐酸氨溴索溶液;
磷酸氢钙二水合物,等等。
以及
2、2022年度国家药品标准提高项目课题目录(通用技术要求)
包括50个中药、化药、生物制品 、理化、微生物等方法学,诸如:
ICH Q4指导原则8个理化分析方法转化;
分析方法建立指导原则的建立;
微生物分析方法验证、确认及转移指导原则,等等。
【上市后监管】
4.12,NMPA印发《药品年度报告管理规定》
NMPA正式印发《药品年度报告管理规定》 ,距离2020年12月征求意见稿 的发布已一年有余。
从监管角度,作为药品监管的“闭环”,上市后药品年度报告制度从形式上规定了企业与监管部门的定期信息沟通。所谓“有法可依,有据可查”,从此,变更 、生产质量管理、药物警戒 ,乃至销售情况,等等全生命周期 的信息,都成为“可查”之“据”。
从实操角度,企业必须为上市后年报制定专门的SOP ,而这个SOP的终点尽管是一份报告,这份报告却可以牵动整个公司上下多个部门、多条流程,改变许多岗位的工作习惯,影响也许相当深远。
识林梳理了中美上市后年报的法规体系,见相关资讯【NMPA药品上市后年报落地,与FDA年报要求的对比梳理】 ,并简要对比了两国年报要求的差异。对于企业读者,多一个角度,对法规的认知和运用也更加充分。
4.15,NMPA印发《药物警戒检查指导原则》
本文为正式指南,征求意见稿 曾发布于2021年12月1日。
所有药企均必读,因此具体内容不在此赘述,仅通过“页面比对”看正式指南与征求意见稿的区别。
这些区别,体现了NMPA结合业界反馈意见的综合考量,有利于进一步理解法规背后的监管思路,更好地开展合规工作。
——检查重点新增“创新药、改良型新药,以及针对儿童、孕产妇等特殊群体使用的药品”,删去了“药品本身存在的固有风险”。 检查资源总是有限的,对于风险尚不够明确的药物,检查自然更加侧重。而所谓“固有风险”,删去的原研可能是难以充分界定。
——需检查的是“药品安全委员会组织结构”,删去了“药物警戒体系组织结构图”。 简化为一个组织结构,更明确。
——查看药物警戒(PV)培训计划、记录和档案,删除了“时间、地点、形式...幻灯视频”之类内容。 作为指南,理应无需如此具体,各企业会有不同的培训形式和记录方式。同理,“记录和数据”部分也删去了许多具体要求。
——PV的SOP 覆盖“关键”PV活动,而非“全部”活动。 “全部”既不必要,也不合理,难以界定。
——对于信号分析评价,新增“查看呈现聚集性信号的病例分析和情况调查资料”。
——提交的核查报告中纳入的安全性信息“包含所有信息来源”。
以上仅为部分示例,这类区别还有许多,识林用户可用“页面比对”工具制作花脸稿,仔细研究。
【政策与监管科学】
4.12,CDE公开2022年部门预算
按照NMPA有关工作要求,CDE依法依规履行公开主体责任和义务,将《国家药品监督管理局药品审评中心2022年部门预算》公开。
与FDA的PDUFA (处方药用户费用法案)或GDUFA (仿制药 用户费用法案)不同,该预算文件并未详细描述预算年度的工作规划,可注意下列几个数据:
——2022年度,预算收入等于预算支出,总计约4亿元(40,182.57 万元)
——其中,“市场监督管理事务”预算37,538.14万元,“药品事务”14,752.03万元,“事业运行”22,567.00万元。
与之相对,FDA2023财年预计申请预算高达84亿美元(见识林资讯:【首仿药180天专营期和加速审批拟修订,FDA申请84亿美元预算用于2023财年工作】 )。其中,与CDE相对应的CDER和CBER,即使不考虑其他职能,只看Human Drugs人用药品部分,2023财年预算也高达22亿美元(见P101,FY2023 President's Budget Request for the FDA)。
【医疗器械】
4.14,NMPA公示《2022年医疗器械行业标准制修订计划项目》
包括:
2022年医疗器械 强制性行业标准制修订计划项目,和
2022年医疗器械推荐性行业标准制修订计划项目。
医疗器械企业可关注自家产品标准是否在修订之列。
【新药批准和报产】
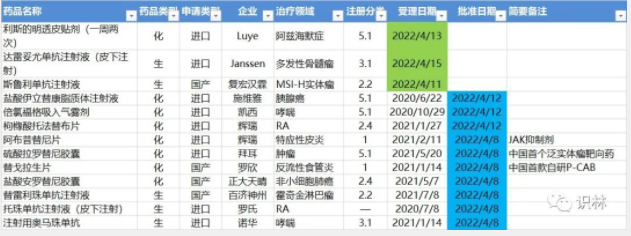
4.11-4.17,NMPA发布10个新药批准,包括国产首款自研P-CAB,CDE受理3个新药上市
注:仅列出新药(包括改良型新药和生物类似药 )的上市申请和批准上市信息。在“以临床价值为导向”的背景下,申请上市以及获批的品种,其适应症 、临床研究 策略、注册路径,都值得业界关注和分析。
作者:识林-实木
识林® 版权所有,未经许可不得转载。
岗位必读建议:
QA:确保年度报告的真实性、准确性,并符合法规要求。 注册:了解年度报告的提交要求和流程,确保按时提交。 研发:参与年度报告中上市后研究及变更管理部分的撰写。 临床:提供上市后风险管理计划和相关数据支持。 文件适用范围:
文件要点总结:
年度报告责任主体 :持有人负责年度报告的真实性和准确性,境外企业需指定境内代理人。年度报告制度 :持有人需建立并实施年度报告制度,包括填报、管理和审批流程。信息收集与报告 :年度报告应包含生产销售、上市后研究、风险管理等信息,并于每年4月30日前提交。监督检查与整改 :药品监督管理部门将对年度报告制度进行检查,不符合规定的需在规定时间内整改。信息保密 :未经持有人同意,不得披露商业秘密或未披露信息,除非涉及国家安全或重大公共利益。以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
药物警戒专员(PV):必须熟悉本指导原则,确保药物警戒活动符合监管要求。 质量保证部门(QA):监督药物警戒体系的合规性,确保缺陷及时整改。 注册部门:在药品注册过程中考虑药物警戒要求,确保注册资料的完整性。 研发部门:在药品研发阶段考虑药物安全性,为药物警戒活动提供科学依据。 文件适用范围:
文件要点总结:
检查原则与目的 :强调药物警戒检查的重要性,督促持有人落实药物警戒主体责任。检查适用情况 :明确了常规检查和有因检查的重点考虑因素,确保检查工作的针对性。检查方式与地点 :规定了现场检查和远程检查的方式,以及检查地点的选择原则。缺陷风险等级 :将检查中发现的缺陷分为严重、主要和一般三个等级,并对风险等级的判定提供了具体标准。评定标准 :根据缺陷的严重程度,明确了检查结论和综合评定结论的分类标准。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
QA(质量保证) :应熟悉本办法,确保药品标准制修订工作符合规定。注册 :需掌握本办法,以便在药品标准制修订研究课题中正确执行注册流程。研发 :应了解本办法,以指导药品标准制修订相关的研究工作。工作建议:
QA :监控药品标准制修订研究课题的执行情况,确保符合管理办法要求。注册 :在药品标准制修订研究课题立项和申报过程中,确保所有步骤符合管理办法规定。研发 :在进行药品标准制修订研究时,依据本办法指导研究工作,确保研究质量和效率。适用范围:
要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。