美国 FDA 局长 Scott Gottlieb 于 3 月 19 日在布鲁金斯学会上透露,FDA 正在准备改写药品 GMP 法规的部分内容来加强对原料药(API)生产工艺变更的监督。根据 FDA 正在起草的对 GMP 法规的修订,制药商必须向 FDA 报告有关对于原料药生产工艺计划变更的更多信息。
去年在缬沙坦原料药中出现的潜在致癌性基因毒性杂质导致了 FDA 决定加强对 API 生产变更监督。FDA 认为杂质产生于六年前批准的缬沙坦 API 主要供应商对 API 生产工艺的变更。GMP 修订是到目前为止 FDA 对于缬沙坦危机的最重要响应。
Gottlieb 表示“这是在很长一段时期内对 GMP 法规的首次真正现代化”。美国 GMP 于 1963 年首次发布,现行 GMP 是 1978 版本,之后虽然有过若干次修订,但都是对文字语句的微小修订,FDA 曾于 2002 年左右准备大修 GMP,但经过一番论证后被否定。他表示,此次修订将“专门针对我们关于 API 生产商的要求,因为这是存在漏洞的地方,不仅仅涉及到潜在风险还可能导致短缺。”他表示,GMP 修订将“对生产商提出更多报告工艺变更的要求,从而我们可以在这些变更落实到位之前获得完整信息,以开展适当的评估。”
当被问及有关 FDA 正在起草的法规的进一步信息时,FDA 新闻办公室提供了一份关于 Gottlieb 对于“生产现代化”评论的声明,表示法规涉及《联邦法典》(CFR)第 21 章 210 和 211 部分的 GMP 法规。声明补充指出,FDA “打算根据需要继续现代化或澄清法规,以与其它 FDA 法规和国际 CGMP 标准协调一致。”新闻办公室拒绝提供更多细节,“因为拟议法规草案目前仍在制定中。”但声明并没有提及 21 CFR 314.70,该法规条款涉及到批准后生产变更的报告和审查。
Gottlieb 还分享了关于缬沙坦问题起因的一些见解,缬沙坦问题同时还影响了同属血管紧张素 II 受体阻断剂(ARB)类别的其它降压药。Gottlieb 表示,“我不认为这是我们不知道发生工艺变更的情况。我认为这是我们不知道这种特殊杂质可以从已发生的工艺变更中引入的情况。”在浙江华海药业缬沙坦 API 中引入亚硝胺杂质的生产工艺“已获得专利并公开发表。很多 ARB 生产商同时都采用了这种生产工艺。”他指出,现在认为生产商通过用溶剂洗涤 API 引入了极低水平、难以检测的亚硝胺杂质。“对于一些生产商而言,从理论上说,他们使用相同的溶剂一遍又一遍的洗涤 API,随时间的推移杂质逐渐增加。”
Gottlieb 还表示,FDA 在过去几年中一直在增加有机化学家的数量,以评估生产变更提案中形成类似危险杂质的风险。“我们有许多有机化学家,他们从根本上评估生产变更以寻找可能通过有机化学变更引入杂质的步骤。”他表示,FDA 药品质量办公室(OPQ)正在通过增加有机化学家,尽其所能减少非预期杂质的形成风险,“这些有机化学家具有研究有机化学中化学反应的专业知识,并能够判断化学反应的变化会如何引入某些风险。”
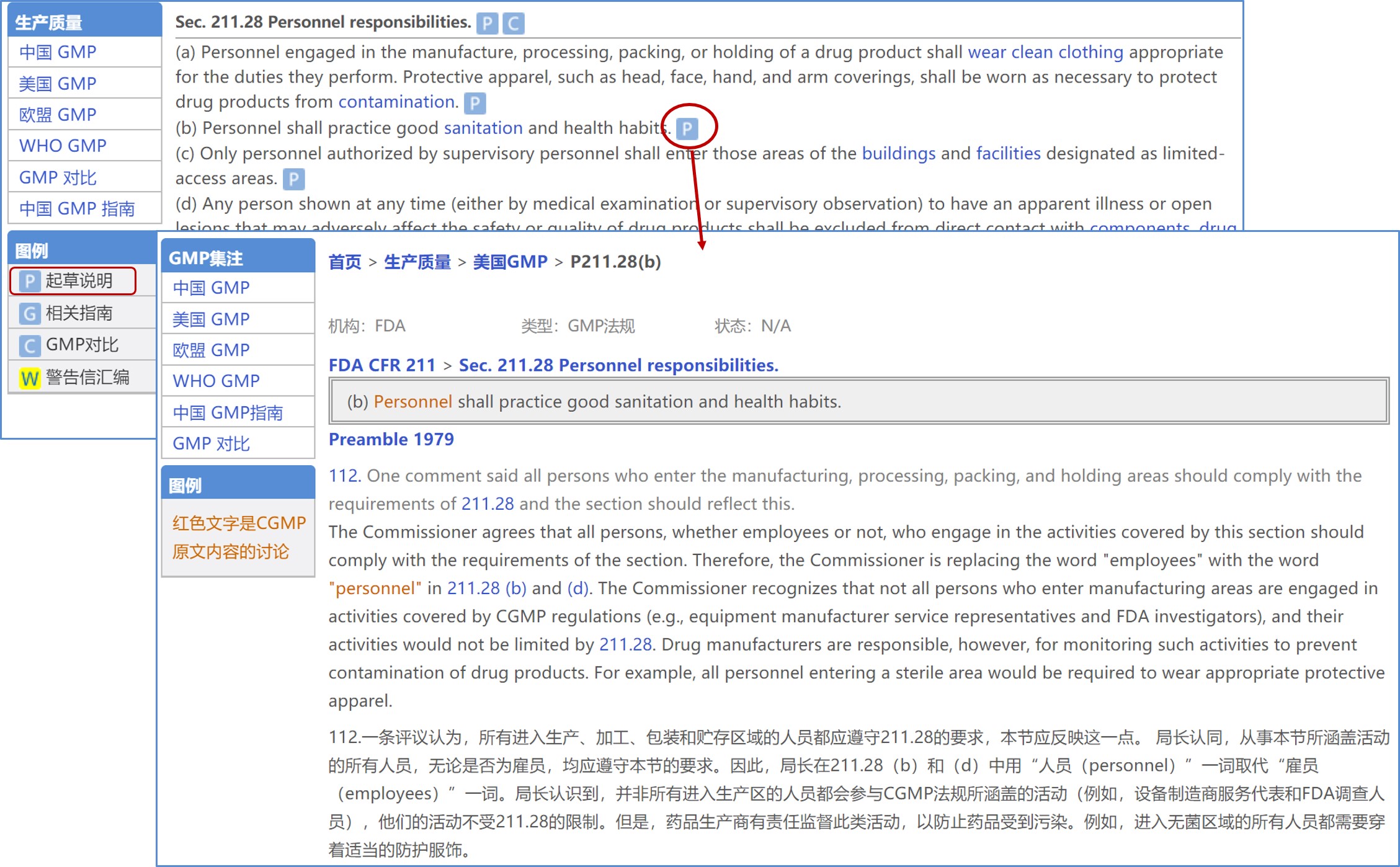
识林知识:起草说明(Preamble)
FDA 在修订 GMP 时通常会在联邦公报征求业界意见,并把官方回应整合汇编成修订的“起草说明”。起草说明有助于更深入了解 FDA 关于法规的监管考虑。起草说明包括法规起草或修订时公众的评论意见和 FDA 局长对于接受或拒绝评论意见的理由 , 代表了 FDA 对具体 GMP 要求的立场,可以在解释 GMP 条款时作为宝贵参考。识林收录并翻译了起草说明 , 并以注释性法规的形式将起草说明中的条目分布到法规的具体条款处,以方便读者学习。