|
首页
>
资讯
>
英国 MHRA 2019 年 GMP 检查缺陷趋势分析
出自识林
英国 MHRA 2019 年 GMP 检查缺陷趋势分析
2020-10-20
英国药品与医疗保健产品监管机构(MHRA)于 2020 年 10 月 15 日发布了其 2019 年的 GMP 检查缺陷。与去年一样,2019 年 GMP 检查缺陷数据同样采用了电子表格的形式,共5293 行数据。有兴趣者可以根据需要自行解析和呈现数据。【英国 MHRA 2018 GMP 检查缺陷趋势分析:数据总览 2020/05/26】
本文将以图表的方式分析呈现一些检查缺陷的分布、趋势以及历年对比,供企业作为自查和持续改进的依据,对照缺陷发现开展自评,以提前发现问题并改进和完善。
检查分布
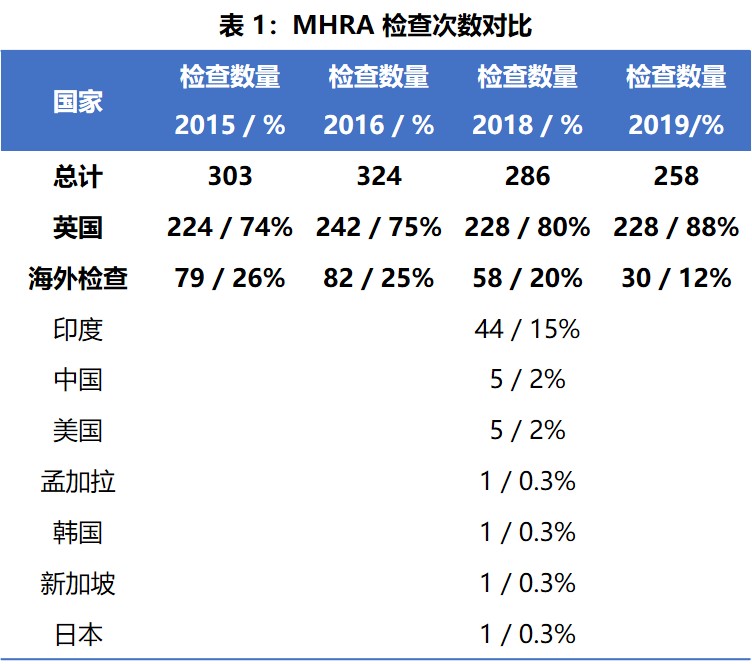
MHRA 在 2019 年对英国国内和国外共执行了 258 次 GMP 检查,其中,英国本土检查 228 次,海外检查 30 次。但是没有像 2018 年那样提供针对海外特定国家/地区的数据。表1 列出了 MHRA 在 2015 年、2016 年、2018 年和 2019 年的检查数量对比。可以看出,与往年一样,几乎大部分检查都在英国国内,且近年来在英国执行的检查百分比持续增加,海外检查的比例持续下降。
GMP 章节和附录
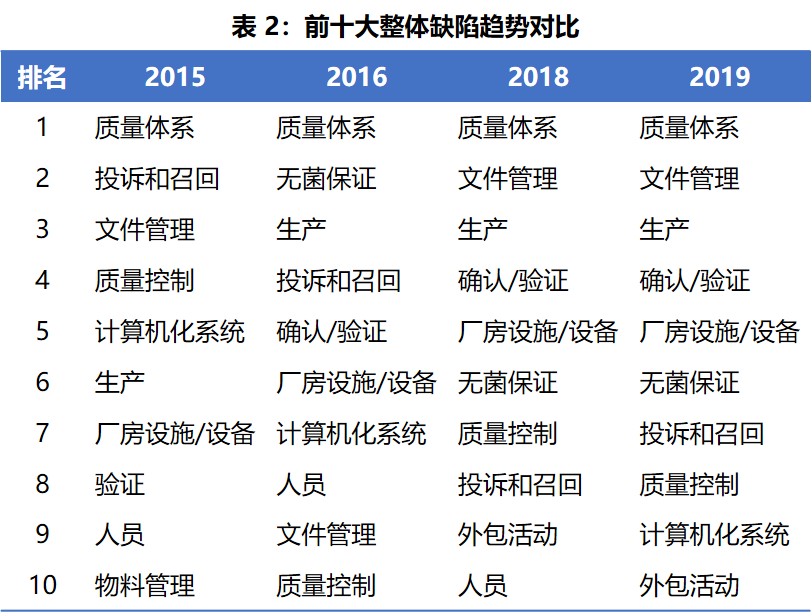
表 2 显示了 2015、2016、2018 和 2019 年所有缺陷中出现最多的欧盟 GMP 章节和附录趋势对比。质量体系四年均位居榜首。2018 年与 2019 年趋势基本一致,前六大常发生缺陷的章节是相同的,没有变化。人员在 2019 年掉出前十,排在第十一位。
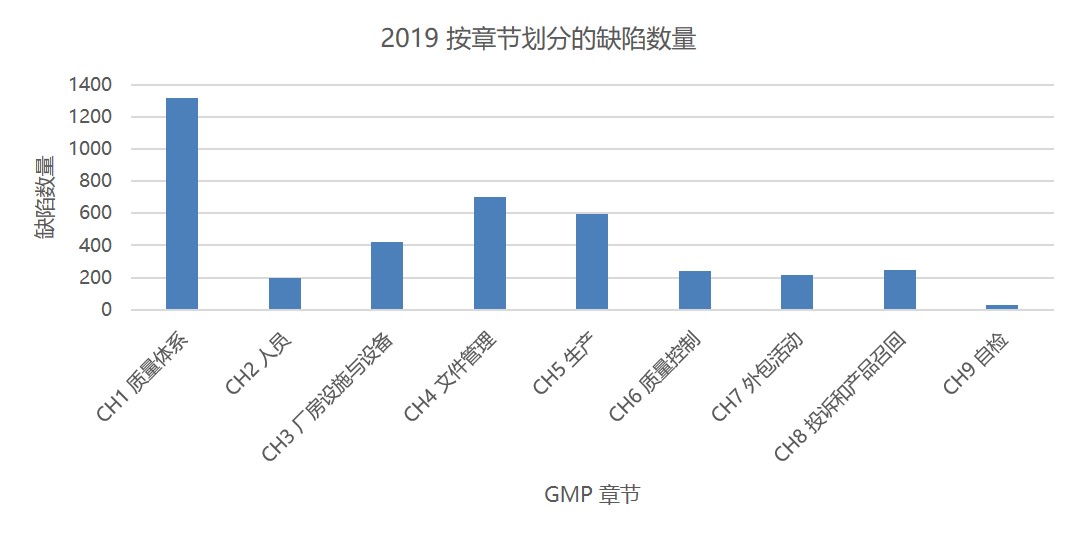
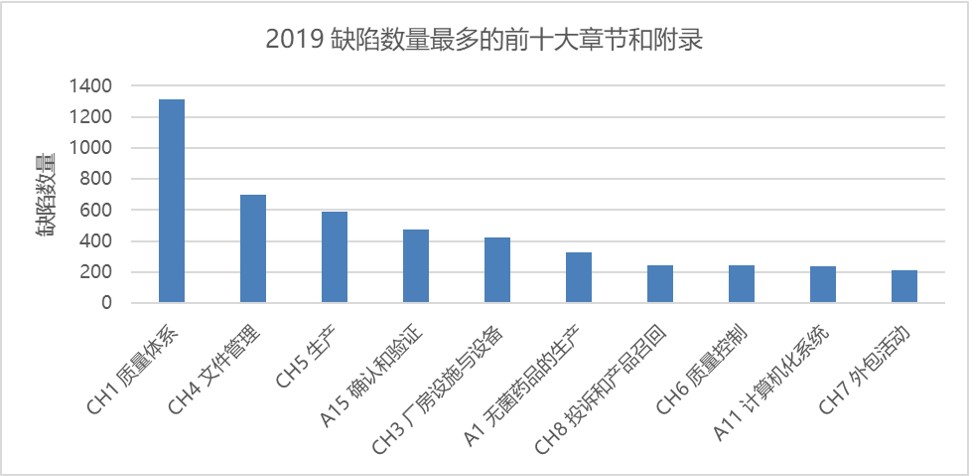
图1 和图2 分别显示了根据所引 GMP 章节或附录的缺陷总数。GMP 第一章质量体系一直以来都是缺陷引证数量最多的章节,2019 被引证缺陷数约 1300 项。其次是文件管理章节(700 项)。然后是第五章生产 593 项缺陷。
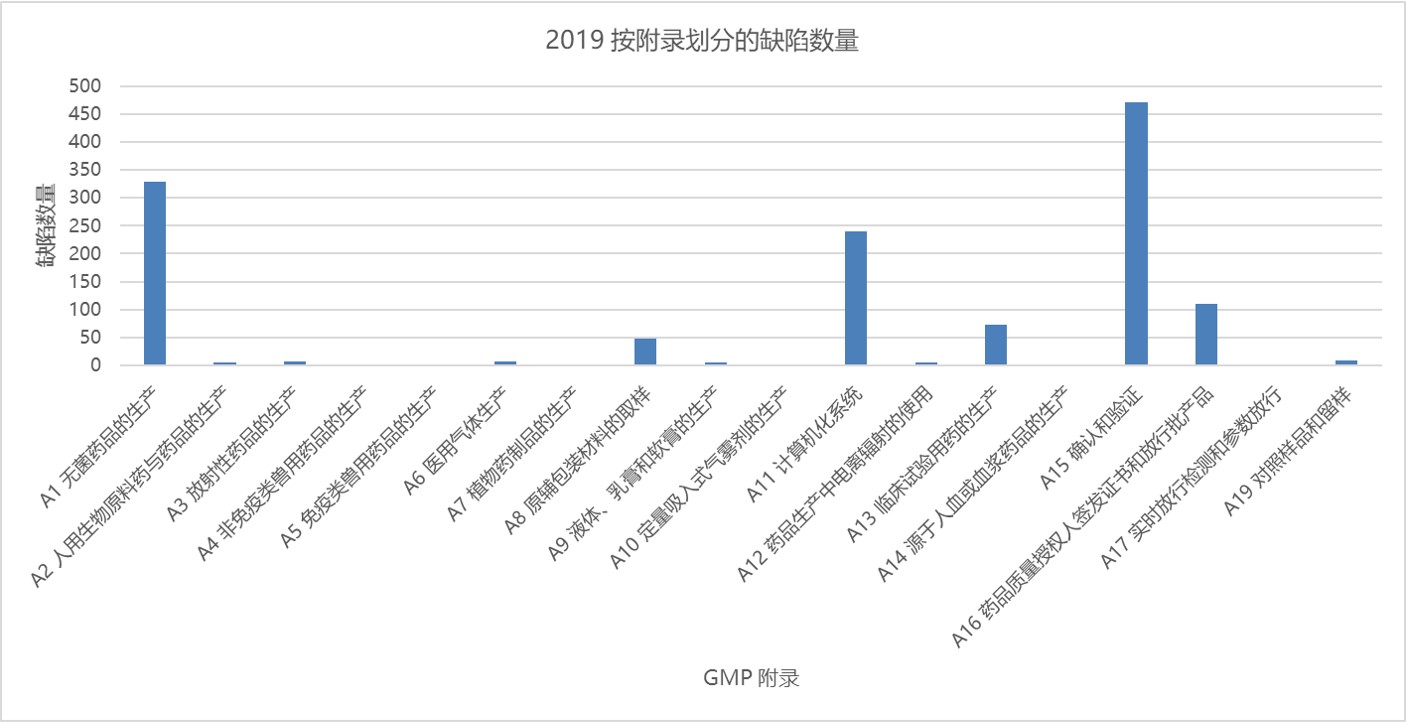
与 2018 年相同,附录 15 确认和验证与附录 1 无菌保证位列第一和第二,分别包括 472 项和 329 项缺陷。然后是附录 11 计算机化系统和附录 16 药品质量授权人签发证书和放行批产品。
图 3 显示了将 GMP 附录和章节合并在一起时位列前十的类别。
严重和主要缺陷
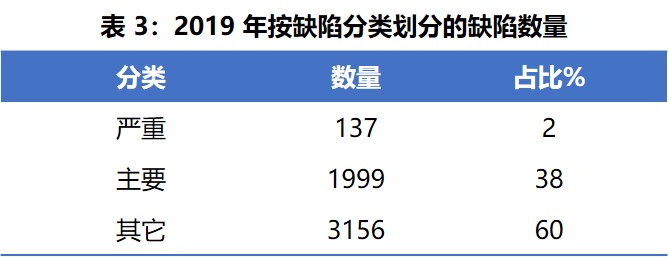
表3 列出了 2019 年按缺陷严重程度分类的情况。严重缺陷占总数的 2%,主要缺陷占 58%,其它缺陷占 60%。
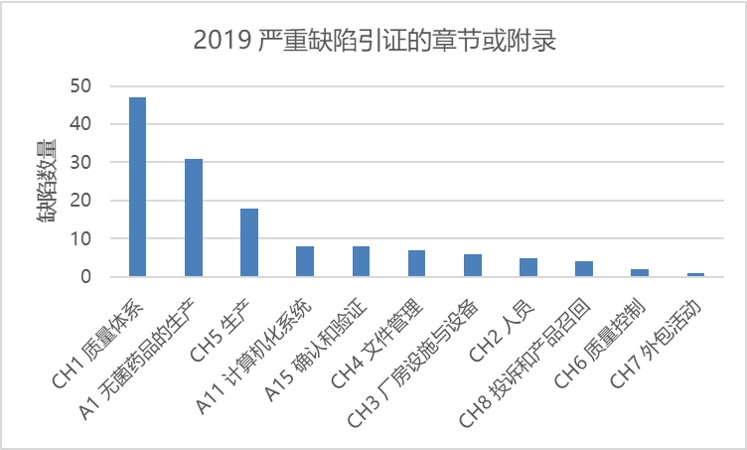
总共 5292 条缺陷中,大多数严重缺陷和主要缺陷都集中在几个章节和附录中。图 4 显示了严重缺陷所在章节或附录及其数量。在严重缺陷中 34% 与质量体系有关,23% 与附录1 无菌保证有关。
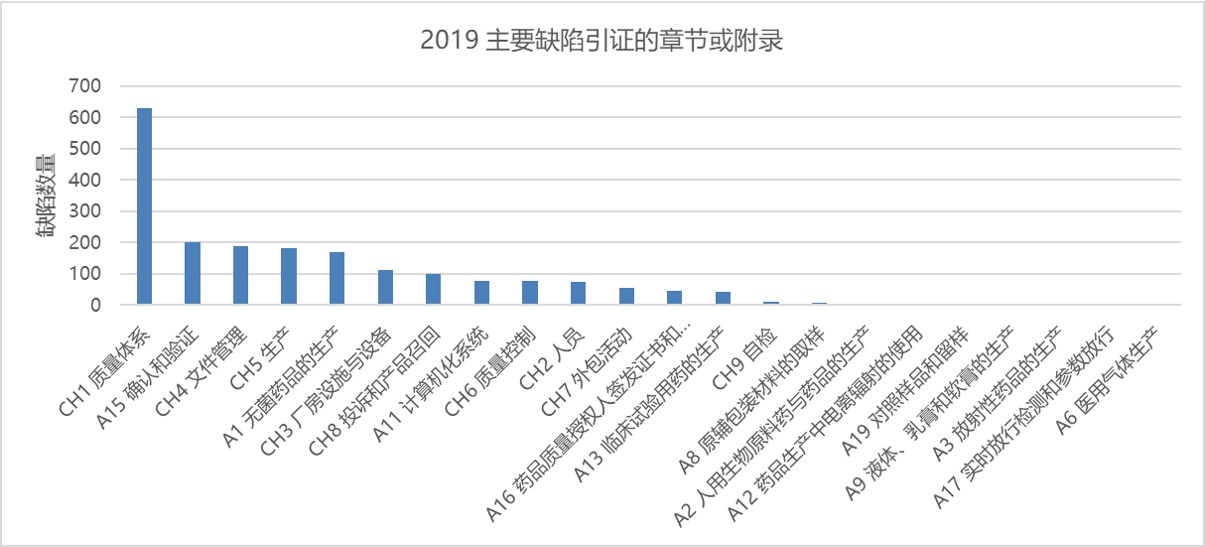
图 5 显示了与主要缺陷相关的章节和附录。无论是所有缺陷、还是严重缺陷或者是主要缺陷,质量体系章节的缺陷引证数量都稳居第一。2018 年也是如此,表明了 MHRA 对完善的质量体系的关注。
作者:识林-椒
识林®版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
适用岗位必读建议: - QA(质量保证):应深入理解附录15的要求,确保质量管理体系与GMP标准一致。
- 生产:必须遵循验证主计划(VMP)和相关文件,确保生产过程中的设备和系统符合GMP要求。
- 工程:在设计和安装阶段应确保设备、设施满足URS,并参与DQ、IQ、OQ和PQ等确认活动。
- 研发:在产品开发阶段应考虑工艺验证的要求,确保研发到生产的顺利过渡。
- 验证:负责制定和执行确认与验证计划,确保所有相关活动符合附录15的指导。
文件适用范围:
本文适用于化学药、生物制品、疫苗和中药等药品类型,包括创新药、仿制药、生物类似药和原料药等注册分类。适用于在欧盟成员国注册和生产的药物,针对Biotech、大型药企、跨国药企、CRO和CDMO等企业类别。 文件要点总结: - 确认与验证原则:强调了确认和验证活动应贯穿药品生命周期,基于质量风险管理。
- 文件记录重要性:确认和验证过程中产生的所有文件应得到适当批准,并明确文件间的相互关系。
- 设备和系统确认:从URS到DQ、IQ、OQ和PQ,每个阶段都有其特定要求和标准。
- 工艺验证方法:介绍了传统验证、同步验证、持续工艺确认和混合方法,并强调了工艺验证的科学性和风险基础。
- 变更控制:变更应通过质量风险管理评估,确保对产品质量的影响得到控制。
以上仅为部分要点,请阅读原文,深入理解监管要求。 岗位必读指南: - 质量受权人(QP):必读。确保对药品生产和放行的全过程有深入理解,并按照附录要求执行认证和放行。
- 生产管理(PM):必读。了解QP的放行要求,确保生产过程符合GMP和MA规定。
- 质量保证(QA):必读。监督和确保QP放行流程的合规性,提供必要的GMP支持。
- 注册部门:必读。理解放行要求,确保注册文件符合规定。
- 研发(R&D):了解。知晓放行流程,确保研发阶段的产品符合放行标准。
工作建议: - QP应持续接受相关产品类型、生产工艺的培训,确保认证工作的准确性和合规性。
- PM需与QP紧密合作,确保生产记录的完整性和可追溯性。
- QA应建立和维护放行流程的审计跟踪,确保所有放行活动符合GMP要求。
- 注册部门应确保所有上市许可文件与放行流程保持一致,及时更新注册信息。
文件适用范围:
本文适用于欧盟内持有上市许可或供出口的人用药品和兽用药品,包括临床试验用药(IMP)。适用于化学药、生物制品等,不包括官方控制机构批放行的血液和免疫产品。发布机构为欧盟,适用于Biotech、大型药企、跨国药企等。 要点总结: - QP认证责任:QP负责确保每批药品的生产和检查符合GMP和MA要求,包括对生产全过程的了解和认证。
- 生产和放行流程:详细规定了认证程序、第三方GMP评估、非预期偏差处理以及批放行的具体要求。
- 多场地生产协调:若生产过程涉及多个场地,QP需确保所有场地的生产活动符合GMP和MA要求,并在认证中考虑其他QP的确认。
- 进口药品的特殊要求:对于在欧盟以外生产的药品,QP需确保满足进口和认证的相关条件,包括活性成分的分析和检验。
- 记录和文档管理:QP认证的记录必须保持更新,并在规定的时间内可供监管机构检查。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |