首页
>
资讯
>
2011-2020年美国和欧洲药品监管审评时长及提交时间差异分析
出自识林
2011-2020年美国和欧洲药品监管审评时长及提交时间差异分析
2023-10-20
药监机构的审评速度通常是制药行业讨论的焦点,2023年10月17日,收载于 《内科学年鉴》(Annals of Internal Medicine)杂志的一篇题为“Regulatory Review Duration and Differences in Submission Times of Drugs in the United States and Europe, 2011 to 2020”的文章中以2011 年至 2020 年在美国、欧洲(欧盟和瑞士)批准的新药为样本评估了首个及补充适应症 的监管审评持续时间以及美欧之间提交时间的差异。其中,从提交到批准的总体中位审评时间(减去时钟停摆时间)在美国为 39 周,在欧盟为 44 周,在瑞士为 44 周。补充适应症的中位总审评时间在美国为 26 周,在欧盟为 40 周。欧盟和瑞士的申请提交时间中位数分别比美国晚 1.3 周和 17.9 周。
样本的获取及分析
美国、欧盟和瑞士,这三个司法管辖区的经济相似。通过美国 (FDA) 和欧盟 (EMA) 的公开数据库以及瑞士 (Swissmedic) 的公开/非公开数据库,研究团队获得分析样本包括2011年1月1日至2020年12月31日期间在美国以及2021年12月31日之前在欧盟和瑞士批准的所有新药。
主要分析队列包括241 种药物,美国有 128 种有补充适应症(总共 331 种),欧盟有 87 种有补充适应症(总共 206 种)。次要分析的研究队列包括 FDA 批准的 391 种新药、 EMA 批准的 310 种新药和 Swissmedic 批准的 266 种新药。
研究的主要结果是审评持续时间减去“时钟停摆”持续时间,次要结果是总审评时间。汇总统计数据(平均值、中位数和IQRs[Interquartile range])用于描述司法管辖区之间和治疗领域之间审评时间的差异。
首个适应症的审评持续时间
向3个机构都提交了申请的药品从提交到批准的总体中位审评时间(扣除时钟停摆时间)在美国为 39 周,在欧盟为 44 周,在瑞士为 44 周(图 1)。
注:该图显示了3 个机构(FDA、EMA 和 Swissmedic)批准的所有药物按疾病领域归类的审评时间,不包含时钟停摆时间。左图显示按机构和疾病领域划分的审核时间分布(箱线图和异常值)。右图显示了各机构之间审评时间未经调整的四分位数和中位数差异。
对比每个药物的审评时间,EMA 比 FDA 平均多3.7 周(IQR,6.7 - 14.9 周),而 Swissmedic 比 FDA 平均多 0.3 周(IQR,10.6 - 15.3 周)。
美国和瑞士中位审评时间最短的是抗肿瘤药物 ,欧盟则是抗感染药物。
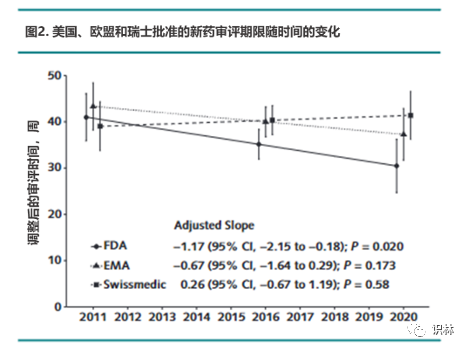
在过去十年中,美国和欧盟的平均审评时间(扣除时钟停摆时间)略有缩短,瑞士的情况恰好相反(图2)。
注:该图显示了根据解剖治疗化学分类(ATC )调整的审评时间的趋势线,提供了这些调整后趋势的数值估计
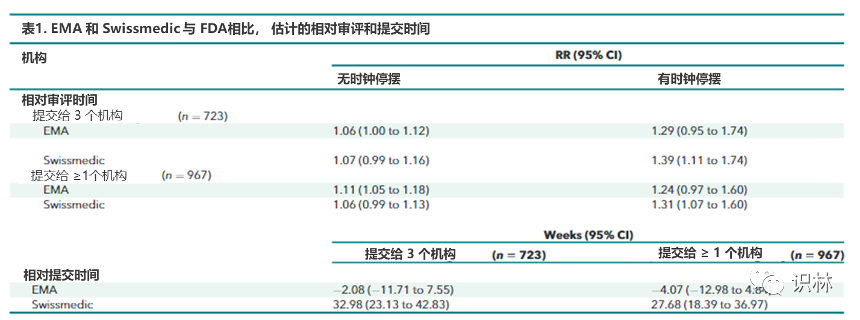
注:RR = relative rate. 顶部显示了机构相对于 FDA 的估计调整后审评时间相对比率(有和没有时钟停摆),并使用提交给所有 3 个机构的药物样本 (n = 723) 和提交给任何机构的所有药品样本 ( n = 967)。底部显示 FDA 与 EMA 和 Swissmedic 之间几周内的估计调整差异。
扣除时钟停摆持续时间,药品从提交直至获得批准的速度EMA比 FDA平均慢 6%(相对率 [RR],1.06 [95% CI,1.00 至 1.12]),而 Swissmedic比FDA平均慢 7%(RR,1.07 [CI,0.99 至 1.16]);
包含时钟停摆持续时间时,提交至获批的速度EMA 比 FDA 慢 29%(RR,1.29 [CI,0.95 至 1.74]),Swissmedic 比 FDA 慢 39%(RR,1.39 [CI,1.11 至 1.74]);(见表1)
大约2/3的在美国获批的新药(158/241)、1/5的于欧盟获批的新药(50/241)以及1/3的在瑞士获批的新药(81/241)都至少获得了1条加速途径的资格。这类药物的审评时间较短:在美国加快 45%(RR,0.55 [CI,0.43 至 0.71]),在欧盟加快 44%(RR,0.56 [CI,0.51 至 0.61]),在瑞士速度快了 42%(RR,0.58 [CI,0.55 至 0.61])(图 3)。
注:a) 呈现扣除时钟停摆时间的回归估计, b) 呈现包含时钟停摆时间的回归估计。该图表示FDA、EMA和Swissmedic当局优先审评 药物(首个适应症 )与非优先药物相比的审评时间相对比例。
补充适应症的审评期限
补充适应症的中位总审评时间在美国为 26 周,在欧盟为 40 周。FDA 和 EMA 的 RR 相似:FDA 补充适应症比首个适应症快 26%(RR,0.74 [CI,0.64 至 0.84]),EMA 快 27%(RR,0.73 [CI,0.84])。0.68 至 0.78])(表 2)。
注:本表列出了补充适应症与原始适应症的审评时间的相对比率。
美国和欧洲首个适应症提交时间的差异
对比新药提交时间的中位数,欧盟比美国晚1.3 周,瑞士比美国晚 17.9 周。使用固定效应模型进行回归分析的结果显示,美国和欧盟的新申请提交时间几乎没有任何差异(-2.1周[CI,-11.7至7.6周]),瑞士的申请提交时间晚于美国(33.0 周 [CI,23.1 至 42.8 周])(见表1)。
总结
扣除“时钟停摆”时间后,美国、欧盟和瑞士的新药审评用时相近。如果考虑“时钟停摆”时间,美国的审评用时比欧盟和瑞士要短很多。这种审评时长的差异可通过美欧实施不同的审评制度来解释。
在 3 个司法管辖区中,补充适应症的审评持续时间比首个适应症要短得多,补充批准在抗癌药物中尤其常见。对补充适应症的更快审评反映的可能是更大的安全数据库或更长时间的药物临床随访减少了审评机构继续深入审评已批准产品的相同临床前和早期试验的需要。
总体而言,申请通常先在美国提交,欧盟稍晚,在瑞士提交最晚。如果美国和欧洲之间的提交时间差异继续最小化,有效的监管审评时间将有助于公众及时获得高价值药物。
作者:识林-白蜡
识林® 版权所有,未经许可不得转载。