|
首页
>
资讯
>
31个省局关于药品上市后变更实施和技术问答分析与探索
出自识林
31个省局关于药品上市后变更实施和技术问答分析与探索
2023-10-12
2023年9月25日,四川省局药品监督管理局发布了上市后变更备案共性问题解答,笔者对国内的《中华人民共和国药品管理法》、《药品注册管理办法》及全国各省/市/自治区药品监督管理局(以下简称“省局”)发布的药品上市后变更管理办法实施细则进行了通读和复习(识林省局变更细则汇总页面),总结以下几点:
1. 在2019年8月发布的《药品管理法》中第七十九条、一百二十四条和一百二十七条提及了药品上市后变更的法律规定。其中第七十九条提及了药品生产过程中的变更,按照其对药品安全性、有效性和质量可控性的风险和产生影响的程度,实行分类管理。属于重大变更的,应当经国务院药品监督管理部门批准,其他变更应当按照国务院药品监督管理部门的规定备案或者报告。第一百二十四条和第一百二十七条着重提及了未经批准进行的重大变更和未按要求进行的备案或年报的处罚规定。
2. 在2020年1月发布的《药品注册管理办法》中第六条、第十一条、第二十九条、第三十九条、第四十条、第七十七条、第七十八条、第七十九条、第八十条、九十六条、一百零六条和一百二十三条提及药品上市后变更的管理和实施。其中第六条说明了不同变更事项的管理负责单位;第二十九条说明了药物临床期间发生变更时申请人应自行评估进行相应的注册申报;第十一条、第三十九条、第四十条、第七十七条、第七十八条、第七十九条和第八十条说明了药品上市后相关的变更评估和注册申报要求;第九十六条明确了药品注册审评时限;第一百零六条说明了药品变更后品种信息更新由信息中心负责;一百二十三条说明了药品批准文号不因上市后的变更而改变。
3. 2021年1月,国家药品监督管理局(NMPA)发布第8号公告《药品上市后变更管理办法(试行)》,随后2021年2月,国家药品审评中心(CDE)发布第15号公告《已上市化学药品药学变更研究技术指导原则(试行)》。上述两个文件是针对药品上市后的技术和法规方面的更细指导和解读。随后各省/市/自治区药品监督管理局在上述两个法规指南基础上陆续发布本省/市/自治区的《药品上市后变更管理类别沟通交流工作程序和要求》和《省局药品上市后变更管理实施细则》。部分省局发布了上面两项中的其中一个,或者两者没有。仅发布了《省局药品上市后变更管理实施细则》有:河南省局、四川省局、甘肃省局、辽宁省局、山西省局、山东省局和贵州省局;仅发布了《药品上市后变更管理类别沟通交流工作程序和要求》有宁夏省局、广西省局和西藏自治区局;天津市局两者都没有发布。如图1所示,除此之外,其他省局发布了上面两项,帮助所在省制药企业更好更快地实施与执行药品上市后变更。发布的时间各有早晚,《药品上市后变更管理类别沟通交流工作程序和要求》多数省局集中在2021年,《省局药品上市后变更管理实施细则》发布较晚的有在2023年。
4. 《药品注册管理办法》规定,中等变更和微小变更由各省局负责相应的注册申报工作。随着各省制药企业的不断发展,技术执行的问题解答越来越迫切,因此全国多个省/市/自治区局发布了各自的上市后变更技术问答和共性问题解答,共有13个局:包括天津市局、云南省局、四川省局、湖北省局、海南省局、陕西省局、辽宁省局、安徽省局、江苏省局、吉林省局、山东省局、重庆市局和内蒙古自治区局,其他18个省局待发布类似的技术问答或指南。
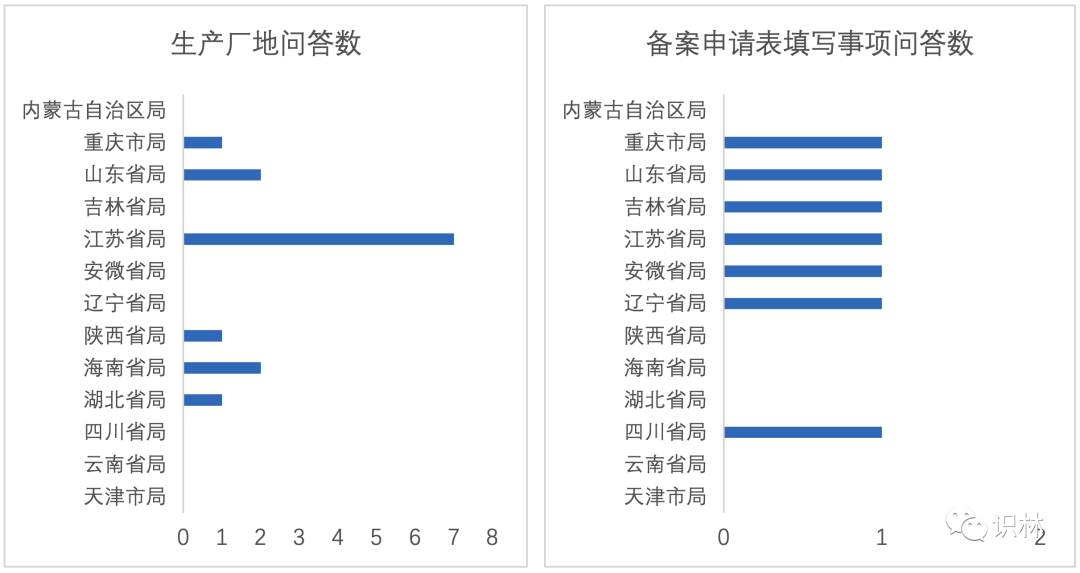
5. 笔者通读各省局的技术问答,其中属江苏省局和山东省局较为完整。各省局发布的技术问答主题较多,笔者总结了常见的十个主题关键词,包括效期延长,原料药变更、生产厂地变更、备案申请表填写事项、说明书/标签修订、溶出曲线对比、工艺验证批次数、稳定性数据、包材变更和关联变更问答。笔者根据以上关键词统计在13个省局的技术问答中出现的数量。如图2。
从图2中可以看出:
- 效期延长、原料药变更和稳定性数据主题基本上在各省局的技术问答中有出现,说明它们是上市后备案中各省局最为需要解决的问题。
- 技术问答次数少不代表所在省局对该技术问题不关注,部分省局会专门针对一个普遍的技术问题发布一个指南,比如天津市局和云南省局针对药品上市后变更效期延长单独发布了指南。
- 十个主题词是笔者根据自身知识总结的,并没有总结所有关键词,还有其它主题词没有纳入。
- 十个主题词中,江苏省局和山东省局出现的次数和问答数位居前列,推测是因为在这两个省份的制药企业较多,面临的技术问题也会相应增加。
- 一些制药大省/市,如浙江、上海和广东没有发布技术问答,其制药企业可通过沟通交流方式解决实际工作中遇到的问题。
本文作者简介:曾文亮,现就职于迪哲医药,从事新药研究与注册工作。
识林®版权所有,未经许可不得转载。
适用岗位及工作建议: - QA(质量保证):必读。需确保变更研究符合指导原则,监督变更实施过程,保证产品质量。
- 注册部门:必读。负责变更注册申请,确保申报材料符合要求。
- 生产部门:必读。根据变更指导原则调整生产工艺,确保生产过程合规。
- 研发部门:必读。参与变更研究工作,提供技术支持。
适用范围:
本文适用于已上市化学药品的药学变更研究,包括化学原料药和化学制剂。适用于不同注册分类的药品,包括创新药和仿制药。由中国药品监督管理局(NMPA)发布,适用于各类企业,包括Biotech、大型药企、跨国药企、CRO和CDMO等。 要点总结: - 变更研究主体责任:持有人/登记企业负责设计并开展变更研究,全面评估变更对药品的影响。
- 变更分类:根据对药品安全性、有效性和质量可控性的影响风险,变更分为重大变更、中等变更、微小变更。
- 关联变更:一项变更可能伴随或引发其他变更,需综合考虑各项变更研究工作的要求。
- 稳定性研究:变更后需进行稳定性研究,必要时增加研究批次或延长研究时间。
- 包装材料和容器变更:变更可能影响药品的保护性、功能性、安全性和质量,需进行相应的研究验证。
以上仅为部分要点,请阅读原文,深入理解监管要求。 适用岗位及工作建议: - QA(质量保证):必读。确保公司变更管理流程符合法规要求,监督变更实施过程。
- RA(注册事务):必读。负责变更申请的提交、审批和备案工作,确保变更文件的合规性。
- 生产管理:必读。负责变更实施的现场管理,确保生产过程符合变更要求。
适用范围:
本文适用于中国境内的化学药、生物制品、疫苗、中药等药品的上市后变更管理,包括创新药、仿制药、生物类似药、原料药等注册分类,适用于Biotech、大型药企、跨国药企、CRO和CDMO等各类企业。 要点总结: - 变更管理责任:强调持有人为变更管理的责任主体,需建立变更控制体系。
- 变更类别:明确审批类、备案类和报告类变更的管理要求,确保变更不影响药品安全性、有效性和质量可控性。
- 持有人变更管理:规定变更持有人后,需保持药品一致性,变更后持有人负责药品全生命周期管理。
- 生产场地变更管理:要求变更生产场地后,药品质量需与原药品一致,重大变更需报批。
- 变更程序和监督管理:规定审批、备案、报告的具体程序,强调监管部门的监督检查职责。
以上仅为部分要点,请阅读原文,深入理解监管要求。 岗位必读建议- 药品注册专员:熟悉药品注册的整个流程和要求,包括临床试验、上市许可、变更和再注册等各个环节。
- 质量保证专员(QA):掌握药品注册过程中的质量标准和质量可控性要求,确保药品注册资料的真实性、一致性。
- 药物警戒专员:了解药品上市后监测和评价的相关规定,负责药品安全性信息的收集和处理。
- 研发部门:关注药品注册管理办法中对药物创新和改良的要求,指导研发方向和策略。
文件适用范围本文适用于中国境内的化学药、中药、生物制品等药品的注册管理,包括创新药、改良型新药、仿制药等注册分类,由国家市场监督管理总局发布,适用于Biotech、大型药企、跨国药企等各类企业。 文件要点总结- 药品注册定义与分类:“药品注册”指申请人依法提出临床试验、上市许可等申请,药品监督管理部门进行安全性、有效性和质量可控性审查的活动。药品按中药、化学药和生物制品等进行分类注册管理。
- 加快上市注册程序:对符合条件的药品注册申请,如突破性治疗药物、附条件批准等,提供加快审评审批的政策支持。
- 关联审评审批制度:“化学原料药、辅料及直接接触药品的包装材料和容器”实行关联审评审批制度,确保药品制剂与相关物料的质量。
- 药品注册核查与检验:为确保药品研制合规性和数据可靠性,开展药品注册核查,并对样品进行标准复核和检验。
- 药品上市后变更与再注册:持有人应主动开展药品上市后研究,对变更事项进行分类管理,并在规定时限内申请药品再注册。
以上仅为部分要点,请阅读原文,深入理解监管要求。 法规指南解读适用岗位(必读)- 药品监督管理部门:全面理解法规要求,执行监督管理职责。
- 药品上市许可持有人:确保药品全生命周期的合规性。
- 药品生产企业:遵守生产质量管理规范,保证药品质量。
- 药品经营企业:遵循经营质量管理规范,确保流通环节合规。
- 医疗机构:合理用药,保障患者用药安全。
- 研发部门:遵循药物研发规范,促进新药创新。
工作建议- 药品监督管理部门:加强法规培训,提升监管能力。
- 药品上市许可持有人:建立风险管理体系,主动报告不良反应。
- 药品生产企业:持续优化生产流程,确保产品质量。
- 药品经营企业:建立严格的药品追溯体系。
- 医疗机构:加强药师培训,提升处方审核能力。
- 研发部门:关注新药研发政策,加快创新步伐。
文件适用范围本文适用于中国境内所有药品类型(中药、化学药、生物制品等),包括创新药、仿制药、原料药等注册分类,针对Biotech、大型药企、跨国药企、CRO和CDMO等企业类别,由全国人民代表大会常务委员会发布。 文件要点总结- 药品研制和注册管理:强调临床价值导向,鼓励新药创新,明确药品注册审批流程和要求。
- 药品生产质量管理:规定药品生产必须符合GMP规范,确保药品质量安全。
- 药品经营和使用监管:要求药品经营企业建立质量管理体系,医疗机构合理用药。
- 药品上市后监管:上市许可持有人需开展风险管理,监测不良反应,确保药品安全有效。
- 法律责任:明确违反药品管理法的法律责任,包括行政处罚和刑事责任。
以上仅为部分要点,请阅读原文,深入理解监管要求。 必读岗位及工作建议: - QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。
- QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。
- 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。
- 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。
适用范围:
本文适用于化学药领域的原料药生产,包括创新药和仿制药,适用于大型药企、跨国药企以及CRO和CDMO等企业类别,发布机构为国际通用标准。 文件要点总结:
原料药的生产质量管理规范强调了从质量管理到生产控制的全过程管理。首先,文件明确了质量管理的原则和机构职责,特别强调了质量保证和质量控制的重要性,并规定了自检、产品质量回顾以及质量风险管理的具体要求。在人员方面,规定了资质、培训和卫生要求,确保员工符合岗位需求。厂房与设施章节详细规定了设计建造、公用设施和特殊隔离要求,以保证生产环境的适宜性。设备章节则涉及设计建造、维护保养、校准和计算机化系统的要求,确保设备运行的可靠性。文件还特别提到了无菌原料药的生产特点,包括生产工艺、厂房设施设备设计、生产过程管理以及环境控制等,这些都是确保原料药质量的关键环节。 以上仅为部分要点,请阅读原文,深入理解监管要求。 |