|
首页
>
资讯
>
FDA分享关于生物类似药开发的经验教训
出自识林
2016-11-02
随着工业界对生物类似药的兴趣持续增长,美国FDA的高级官员敦促申办人很好的利用FDA的建议并遵循最佳实践,以确保其生物类似药开发项目的及时成功。根据FDA新药办公室治疗生物制品副主任Leah Christl表示,目前在美国有针对20个参照产品的66个生物类似产品在研,其中有来自7个不同申办人的10个产品公开宣布向FDA提交了351(k)申请。
生物类似药开发
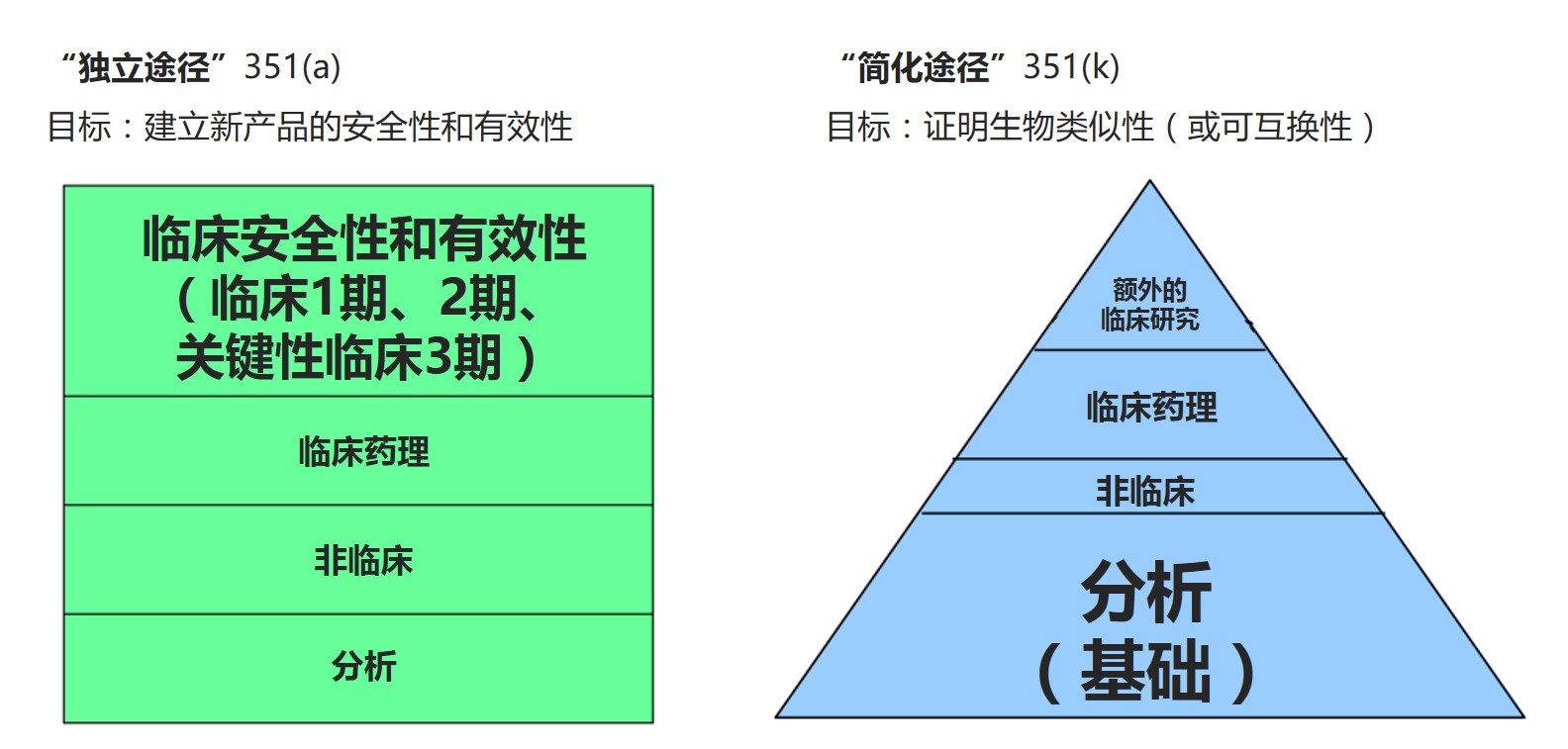
10月27日在美国华盛顿特区举行的药物信息协会(DIA)生物类似药会议上,John Jenkins的报告提醒与会者,生物类似药的主要优点是通过简化途径获得批准。然而,Jenkins表示,生物类似药开发者有时忽略了351(k)的简化属性,并最终做了比所必要的更多的工作。一些申办人难以将生物类似药开发与传统药物开发区分开来:“生物类似药的本意是一个简化程序,不应该复制参照产品开展的每个研究。”
Jenkins表示,对于生物类似药,批准的基础建立在稳健的分析数据证明参照分子和生物类似分子之间的相似性。“这一概念对于利益攸关者理解生物类似药是非常有挑战性的。生物类似药外推是有关基于所有可用数据将从对参考产品所了解的知识外推到生物类似产品。而不是从对生物类似产品研究所了解的适应症外推到其它适应症。这种缺乏对外推的理解会产生重大后果。”
例如,申办人可能最终在证明相似性的必要临床研究之外开展了不必要的比较性临床研究。Jenkins表示,对于患者、医疗服务提供者和支付者来说,这些额外的研究可能导致推迟对生物类似药的获得。
由于更强调生物类似药的分析数据,Jenkins表示,申办人应寻求开发新的创新方法来研究他们的产品。“生物类似药开发项目很复杂,通常涉及试图证明生物类似性的创新方法,我们实际上鼓励使用新方法和研究设计,例如不同的重点、不同的患者群体,增加检测潜在差异的灵敏度以支持证明生物类似性。”
然而,Jenkins指出,这需要花费FDA的大量努力来审评这些新方法。“例如,一个生物类似药项目可能主要集中在正常志愿者中的药代动力学和药效学(PK/PD)的比较,而不是在临床试验中的比较。但这些新方法必须经过合理证明并通过充分的数据和信息支持,这要求广泛的FDA内部科学、监管和法律讨论以达成一致可以支持这样的新项目,这些都非常耗费FDA的资源和时间。”
会议和建议
Jenkins还鼓励开发者利用FDA提供的资源,例如与FDA的正式会议。FDA目前向生物类似药开发者提供五种不同类型的会议以与FDA讨论他们的产品:生物类似药独有的初始咨询会议,以及面向所有生物制品开发者的四类生物制品开发会议(BPD Types 1-4)。
Jenkins表示,“不完整的申请会浪费FDA的资源,并导致开发商和开发计划的延迟。”他补充指出,企业应确保他们对会议的安排以便有足够的时间采纳FDA的建议行动。“我们曾见到过这样的情形,企业与FDA开了BPD Type4会议,转身很快就提交了他们的申请,甚至在他们拿到会议记录之前。”
最后,Jenkins表示,企业在提交申请时需要做好接受检查的准备。“在提交申请时确保你的设施已经准备好接受检查。当我们去检查时,设施需要在生产商业化产品,以便我们可以执行检查。不是别人的产品,不是别人的单克隆抗体,而是属于你的申请的单克隆抗体。”
全球开发考虑
Christl表示,申办人应在全球监管环境下筹划其生物类似药开发项目以最大化回报。
更重要的是,企业应从他们计划在早期提交的所有监管机构寻求建议。“在特定阶段的反馈的重要性可能有一些不同,但如果你想支持一个全球开发项目,你必须考虑的一个事实是,如果你将要在欧盟寻求当前的批准,三到四年内寻求FDA批准……但你正在做开发项目的工作时没有跟FDA沟通,你失去了最大限度的取得数据的机会。”
虽然在许多主要市场的生物类似药监管存在许多相似,Christl告诫指出,企业从监管机构获得的科学建议并不总是相匹配的。在这些情况下,Christal表示,企业应有一个计划来解决这些差异。“如果你没有获得建议并将其整合进开发项目中,将增加你不得不提供额外数据的风险”。
整理:识林-苜蓿
识林®www.shilinx.com版权所有,未经许可不得转载。如需使用请联系admin@shilinx.com
参考资料
|