首页
>
资讯
>
FDA 官方开展的仿制药研究课题概要:口服制剂
出自识林
FDA 官方开展的仿制药研究课题概要:口服制剂
历史版本(Version List) 展开 ∨ 收起 ∧
2025-09-26
在《2024财年GDUFA科学与研究报告》 中,FDA利用内部资源和外部合作,在九大领域开展了超过70项研究课题,涵盖当前仿制药 研发、审评以及监管科学 的重大课题,旨在全面促进仿制药产品开发和可及。
如今的仿制药开发已不同以往,在质量和成本的双重压力下,我国仿制药企业有必要迎头赶上,走在技术的最前沿,同时也要立足中美市场并走向全球。FDA的研究能够提供与一般指南不一样的独特角度,不仅提示开发方向,也可解决实际问题。
识林将逐一编译报告内容,包括代表性课题内容、课题目录以及已发表文献清单,陆续发布,供我国仿制药企业查阅参考。
第六章:口服制剂 在GDUFA III 期间,口服制剂领域研究侧重于:理解口服药品中的成分如何调节生物利用度 ;改进生物相关的溶出方法以及计算机模型,以支持在ICH M13A2 和ICH M13B 下扩展生物豁免和全球协调;开展内部研究,寻求以数据驱动的方式协调窄治疗指数(NTI)药物 的BE标准,以支持未来在ICH M13C 下的协调;以及获取支持调释(MR)口服制剂未来协调所需的数据。
这包括研究以支持对于处方 差异大于FDA指南当前建议的口服速释(IR)制剂实施基于生物药剂学分类系统(BCS) 的BE豁免的可行性,还包括探索处方设计特征对药品体内药代动力学 (PK)的影响,以及研究当MR口服制剂与软质食物同服时,如何使用体外方法学来支持BE。其他研究重点包括开发体内BE研究设计,以维持受试者 安全并确保在特定人群(例如,儿科或老年患者)中的BE。下文简要介绍2024财年相关研究。
生物等效性方法与分析,IR和MR口服制剂,以及人体受试者安全
2024财年多项研究资助的预期成果包括:降低高风险IR药品治疗等效性失败模式;建立以患者为中心的质量标准 ;支持批准MR药品的额外规格;以及评估预测BE结果的体外测试方法。此外,在合同研究75F40121C00020和75F40120C00200以及资助U01FD007959-01下,利用基于生理的生物药剂学/药代动力学(PBBM/PBPK)建模来阐明内在和外在变量(例如,处方设计或食物)对BE评估的影响(涉及PBBM/PBPK建模的研究将在第7章进一步讨论)。
体外分析指导处方开发
资助U01FD007959下的研究旨在确定MR产品的关键质量属性(CQA) 和设计特征,并确定开发MR片剂处方其他规格(缩放因子)的适当因素。该研究使用富马酸喹硫平缓释片作为模型药物,开发了比较表征和溶出方法学,利用表面溶出成像系统来确定药物释放机制和体外溶出度。研究人员采用质量源于设计(QbD) 方法进行处方开发 ,即通过识别和控制CQA来实现理想的质量产品特性。
为了研究辅料 对IR非索非那定口服吸收的影响(资助U01FD005978),选择了两种剂量水平(3 mg和30 mg)的十二烷基硫酸钠(SLS)。SLS是有机阴离子转运多肽2B1(OATP2B1)的抑制剂,通常与非索非那定(OATP2B1的底物)共同处方。该临床研究 于2024财年启动,12名受试者中已有5名完成研究。
在资助U01FD008305中,德克萨斯农工大学健康科学中心旨在确定特定MR产品发生乙醇诱导的剂量倾泻(Alcohol Dose Dumping,ADD)与药物和处方相关的因素,作为改进和支持制定针对ADD评估的MR药品产品特定指南(PSG)工作的一部分。
前沿表征技术:智能药丸,仿生机器食管,计算机预测
在合同研究75F40120C00200下研究了建立以患者为中心的质量标准的生物预测性方法。该项目使用培养管测量格列吡嗪缓释片在不同胃肠区域的体内药物释放速率,用智能药丸记录胃肠道生理条件(例如pH值 和压力),并分析药物血浆浓度。该研究的预期结果将阐明体外差异如何与体内吸收变异相关联。
合同研究75F40121C00132于2024财年完成,并确定机器人软食管(Robotic Soft Esophagus,RoSE)和食道通过时间(Esophageal Transit Time,ETT)分别作为评估口固制剂可吞咽性的工具和指标。研究人员首先建立了适用于在RoSE中对口固制剂进行仿生吞咽的实验方案。然后在RoSE中预定义的波长和速度设置下,测试了不同尺寸、形状和重量的口固制剂的ETT。RoSE测得的口固制剂的ETT对速度变化敏感,但对波长变化不敏感。可能需要在不同速度设置下进行测试,以区分尺寸、形状和重量不同的口固制剂的ETT差异。使用机器学习模型探索了观察到的ETT与产品物理属性之间的关系,但需要更多具有不同物理属性的数据来建立稳健的模型。
在进食条件下,处方变量可能影响口服产品的体内性能(合同研究75F40121C00020)。使用能够模拟机械压力、流体动力应力和食物诱导粘度的生物相关崩解 和溶出方法,来确定在模拟进食条件下辅料 对崩解和溶出的影响。研究团队结合了体外结果和PK数据建立PBPK模型以预测食物效应 。
2024财年FDA还授予了两项新的资助。授予Simulations Plus, Inc.的资助U01FD008388计划使用生物预测性体外方法(即TinyTIM)来预测食物和抑酸剂对无定形固体 分散体制剂吸收的影响。
针对IR产品空腹和进食条件BE必要性的评估
为支持ICH M13A 的开发,FDA进行了内部研究,调查美国FDA批准的IR口服制剂现状,并通过审查美国FDA和欧洲EMA的PSG来评估和比较当前的BE建议。监管机构之间最显著的差异在于BE研究是否需要同时在空腹和进食条件下进行,还是仅需在一种条件下(空腹或进食)进行。ICH 于2024年7月通过的ICH M13A建议基于风险来决定是否需要同时在空腹和进食条件下进行BE研究。FDA的研究工作有助于理解受ICH M13A影响的美国市场IR固体口服制剂 的范围。此外,研究支持FDA修订超过800份PSG以移除关于在空腹或进食条件下进行BE研究的建议,推动ICH M13A实施。协调一致的BE建议简化了仿制药开发,并改善了全球患者对仿制药的可及性。
研究亮点:优化口固制剂可吞咽性评估设计
在2024财年期间,FDA对于当前用于临床可吞咽性评估(CSA)设计的研究成果发表在科学文献上。
口固制剂的可吞咽性是确保用药安全和患者依从性 以防止最终用户不当操作的关键属性,目前尚无最佳实践或明确指南。CSA方法学的差异会影响可吞咽性的测量,进而影响监管决策。因此,迫切需要开发标准化的且经过验证 的评估工具,以产生高质量、具有临床相关性的可吞咽性数据。这不仅将使新药申请人受益,也可用于拟议仿制口固制剂的物理属性偏离监管建议的情形,特别是对于儿科药品。
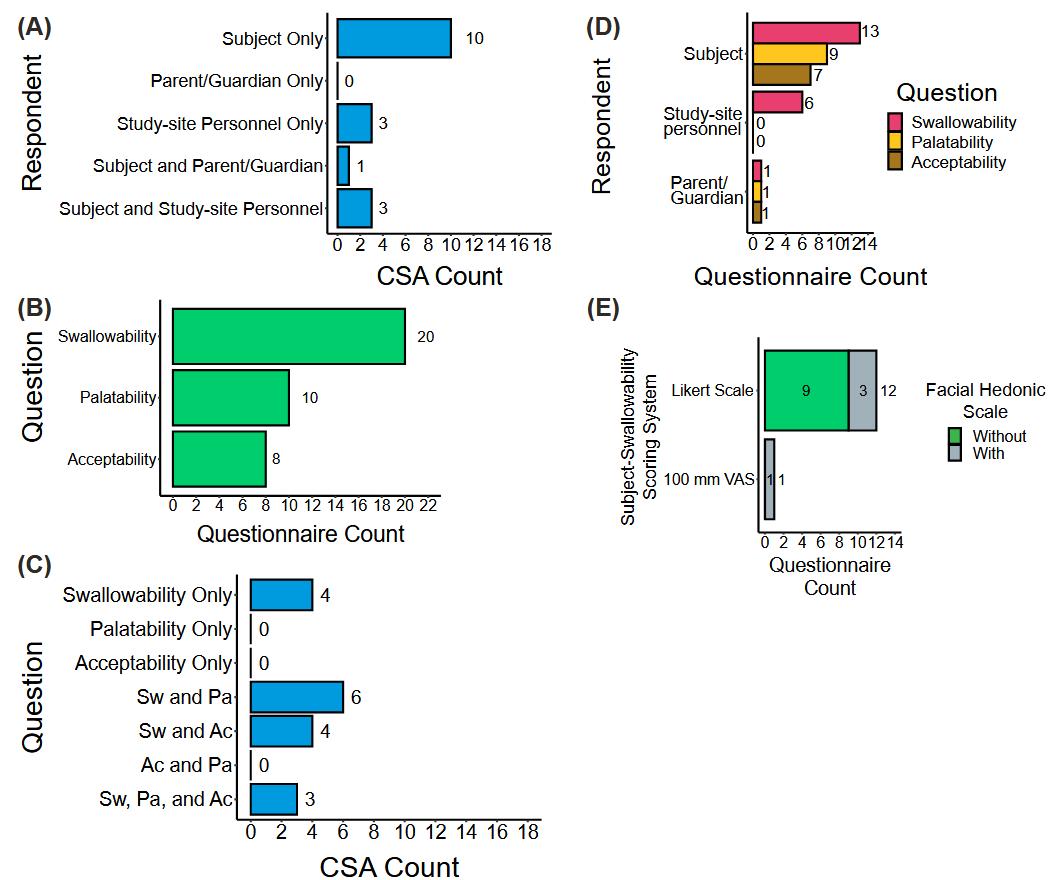
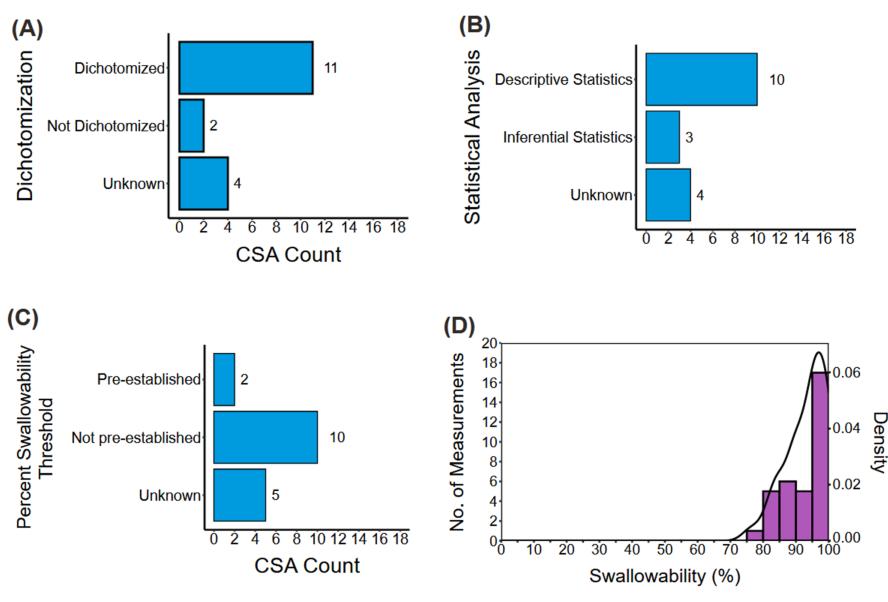
该研究发现CSA研究设计趋势伴随着方法学的巨大差异。在76%的CSA中将可吞咽性作为非主要终点进行评估,这与缺乏足够效力来稳健测量可吞咽性有关。此外,参加CSA的受试者中有76%是儿科患者,这可能是由于申请人需要遵守《儿科研究公平法案》 ,以确定适合该人群的处方。尽管评估可吞咽性的方法多种多样,但总会用到问卷调查,其因受访者、问题类型和回答评分系统的不同而异(图1)。CSA的数据分析方法较为直接,主要使用二分法评分系统和描述性统计,没有预先设定的可吞咽性百分比阈值(图2)。鉴于CSA方法学的差异,不经过大量标准化处理,无法交叉比较可吞咽性结果。
研究项目清单及成果如下(翻译仅供参考):
研究进展与成果
研究项目
New Grants and Contracts
新的资助与项目
Grant (U01FD008305) Factors Related to Drug and Formulation Affecting Alcohol Dose Dumping in Modified Release Oral Drug Products
资助(U01FD008305):影响口服调释制剂发生乙醇剂量倾泻的药物特性与制剂因素
Grant (U01FD008388) From Bench to Bioequivalence: In Vitro Mechanistic Understanding of ASD Drug Products in Simulated Gastrointestinal Conditions
资助(U01FD008388):从实验室到生物等效性 :在模拟胃肠道条件下对ASD药物的体外机制理解
Continuing Grants and Contracts
持续性资助与项目
Grant (U01FD007959) Evaluation of Oral Modified-Release Tablets to Support the Approval of Additional Strengths
资助(U01FD007959):评估口服调释片剂 以支持新增规格的批准
Contract (75F40121C00020) Disintegration and Dissolution of Solid Dosage Forms and Influence of Food Induced Viscosity on its Kinetics, Tools and Methodologies for Bioequivalence and Substitutability Evaluation
项目(75F40121C00020):固体剂型的崩解与溶出及食物引起的粘度对其动力学的影响:生物等效性和替代性评估的工具与方法
Contract (75F40120C00200) Setting Patient-Centric Quality Standards (PCQS) for Modified Release (MR) Oral Drug Products with Biopredictive In Vitro Dissolution-Models
项目(75F40120C00200):基于生物预测性体外溶出模型建立患者为中心的口服调释制剂质量标准
Grant (U01FD005978-04S3) The Effect of Sodium Lauryl Sulfate on the Oral Absorption of Fexofenadine in Humans
资助(U01FD005978-04S3):十二烷基硫酸钠对人非索非那定口服吸收的影响
Completed Grants and Contracts
已完成的资助与项目
Contract (75F40121C00132) Applying a Robotic Soft Esophagus (Rose) to Assess the Swallowability of Opioid Drugs
项目(75F40121C00132):应用机器人软食道(Rose)评估阿片类药物 的吞咽性
Active FDA Research
正在进行的FDA研究
Analysis of the Predictability of Bioequivalence in the Fed State
餐后状态下生物等效性可预测性分析
Baseline Correction in Bioequivalence Studies for Drug Products Containing an Endogenous Compound
生物等效性研究中含内源性化合物药品的基线校正
Development of a Decision Making Procedure in Relation to Male Reproductive Toxicity for the Selection of Bioequivalence Study Population
建立男性生殖毒性 决策程序以选择生物等效性研究人群
Data Mining Hypoglycemic Safety Events in Clinical Trials Involving Combination and Single Products for Treatment of Type 2 Diabetes
在治疗2型糖尿病的联合和单一药物的临床试验中挖掘低血糖安全性事件数据
Development of New Approaches to BE Evaluations of Multi-Strength MR Products
多规格调释制剂生物等效性评价新方法开发
Establish the Framework for Partial AUC Recommendations
建立部分AUC 推荐框架
Evaluation of BCS Class 3 Waiver Expansion
BCS III类药物生物豁免适用性评估及扩展策略研究
Evaluation of the Need for Sprinkle BE Studies
评估撒布性药物BE研究的必要性
Exploration of Food Conditions in Bioequivalence Studies with Pharmacokinetic Endpoints Enrolling Patients in Generic Drug Development
仿制药 开发中基于药代动力学终点的患者人群生物等效性(BE)研究食物条件探索
GDUFA III Product Specific Guidance Improvement for Oral Product
GDUFA III口服制剂产品特定指南改进
Improvement of Drug Dissolution Method for Application to Nanocrystal Drugs
纳米晶药物溶出方法改进
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use - M13A
ICH-M13A
Investigation of Bayesian Estimation Based Procedure for Bioequivalence Assessment
基于贝叶斯估计的生物等效性评估的研究
Modeling and Simulation to Support the Regulatory Harmonization on Bioequivalence Studies for Modified-Release Products
通过建模与模拟支持调释制剂生物等效性研究的监管协调
Qualitative Sameness and Quantitative Sameness/ Similarity (Q1Q2) for Oral Drug Products
定性相同性与定量相同性/相似性(Q1Q2)用于口服药物产品
Reassessment of REMS Recommendations in PSGs for Generic Drug Development
对仿制药开发中PSG的风险评估与减轻策略(REMS) 建议进行重新评估
Safety Considerations for the Selection of Patients as the Subject Population in Bioequivalence Studies with Pharmacokinetic Endpoints for Generic Drug Development
在仿制药开发中以药代动力学 终点进行生物等效性研究时选择患者作为研究对象人群的安全性考量
U.S. FDA Efforts to Support Harmonization of Generic Drug Approved Standards
FDA支持仿制药批准标准协调的努力
Utilization of Pharmacogenomic Information for Subject Recruitment in Bioequivalence Studies
在生物等效性研究中利用药物基因组学 信息进行受试者 招募
Articles
文章
Relative Performance of Volume of Distribution Prediction Methods for Lipophilic Drugs with Uncertainty in LogP Value
分布容积预测方法在LogP值存在不确定性情况下对脂溶性药物的相对表现
Global Harmonization of Immediate-Release Solid Oral Drug Product Bioequivalence Recommendations and the Impact on Generic Drug Development
速释口服固体制剂生物等效性建议的全球协调及其对仿制药开发的影响
Effect of Antioxidants in Medicinal Products on Intestinal Drug Transporters
抗氧化剂在药物制剂中对肠道药物转运蛋白的影响
Antioxidants had No Effects on the In-Vitro Permeability of BCS III Model Drug Substances
抗氧化剂对BCS III类模型药物物质的体外渗透性无影响
Designs of Clinical Swallowability Assessments of Solid Oral Dosage Forms in Regulatory Submissions
在监管申报中固体口服制剂临床吞咽评估的设计
In Vitro Lipolysis Model to Predict Food Effect of Poorly Water-Soluble Drugs Itraconazole, Rivaroxaban, and Ritonavir
体外脂解模型预测难溶于水药物伊曲康唑、利伐沙班和利托那韦的食物效应
Effect of the Similarity of Formulations and Excipients of Approved Generic Drug Products on In Vivo Bioequivalence for Putative Biopharmaceutics Classification System Class III Drugs
对于推定生物药剂分类系统III类药物,已批准仿制药的处方和辅料 相似性对体内生物等效性的影响
Nitrosamine Mitigation: NDMA Impurity Formation and its Inhibition in Metformin Hydrochloride Tablets
应对亚硝胺 :二甲基亚硝胺(NDMA)杂质 的形成及其在盐酸二甲双胍片中的抑制
Characterizing a Design Space for a Twin-Screw Wet Granulation Process: a Case Study of Extended-Release Tablets
表征双螺杆湿法制粒 工艺设计空间 :一种缓释片的案例研究
Using Mechanistic Modeling Approaches to Support BE Assessments for Oral Products
使用机械模型方法支持口服产品的BE评估
识林-梓
识林® 版权所有,未经许可不得转载
必读岗位:
RA(注册):负责提交儿科研究评估,申请延期或豁免,以及处理标签变更。 QA(质量保证):确保儿科研究的执行和报告符合法规要求。 研发:在药物开发过程中考虑儿科适用性,参与儿科研究的设计和执行。 临床:负责实施儿科临床研究,收集和报告数据。 工作建议:
RA:及时提交儿科研究评估,申请必要的延期或豁免,并处理任何标签变更。 QA:监督儿科研究的执行,确保所有活动符合法规要求。 研发:在药物设计阶段考虑儿科剂量和配方,与RA紧密合作以满足法规要求。 临床:设计和执行儿科临床研究,确保数据的准确性和完整性。 适用范围:
要点总结:
儿科研究要求: 规定了新药和生物制品在提交上市申请时必须附带儿科研究评估,以评估药物在不同儿科亚群中的安全性和有效性。数据外推: 在成人和儿童疾病进程及药物效应足够相似的情况下,可从成人研究中外推儿科疗效。延期和豁免: 在特定情况下,FDA可以推迟或豁免某些儿科研究要求,但需提供合理理由和计划。标签变更: 儿科研究结果必须在药物标签中反映,包括任何豁免信息。信息公开: 所有儿科研究评估、延期、豁免和标签变更信息需公开,以提高透明度。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
生物等效性研究人员:深入理解并应用本指南进行生物等效性研究设计和数据分析,确保研究的科学性和合规性。 临床研究部门:在设计临床试验时,考虑本指南对受试者选择、研究设计、样本量计算等方面的要求。 质量管理(QA):确保生物等效性研究的执行和记录符合本指南的规定。 注册部门:在药品注册过程中,依据本指南准备和提交生物等效性研究资料。 文件适用范围:
要点总结:
研究目的与背景 :明确了生物等效性研究的目的,即通过药代动力学终点评估口服固体制剂的生物等效性,并强调了数据完整性的重要性。研究设计原则 :推荐使用随机、单剂量、交叉研究设计,并详细说明了研究人群选择、样本量计算、研究设计和条件等关键要素。数据与统计分析 :强调了非重复研究设计的数据呈现和统计分析方法,包括生物等效性分析人群的考虑、数据呈现、统计分析的一般考虑等。特殊主题 :讨论了内源性化合物、其他速释剂型(如口腔崩解片、咀嚼片、口服悬浮液)、固定剂量组合和pH依赖性等特殊主题。文档记录 :要求生物等效性研究报告应包含完整的研究协议、执行和评估记录,并符合ICH E3的格式。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议:
注册(RA) :必读。需熟悉ICH M13B指南,以便在药品注册申请中正确应用生物等效性豁免标准,与监管机构沟通时准确表述。研发(R&D) :必读。在开发口服固体常释制剂时,应遵循ICH M13B指南,确保产品符合生物等效性要求。质量管理(QA) :必读。需监督生产过程和产品质量,确保符合ICH M13B指南中关于生物等效性的要求。适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA:确保口服固体制剂的生产过程符合本指南要求。 生产:遵循生产管理章节的指导,确保生产过程的合规性。 研发:在设计和选型设备时,参考本指南以确保设备符合生产需求。 临床:在产品实现和验证阶段,确保临床试验用药品的质量符合要求。 文件适用范围:
文件要点总结:
质量风险管理 :强调了质量风险管理在口服固体制剂生产中的重要性,包括原则和工具的应用。生产管理 :明确了生产过程中的关键控制项目,如批次管理和清场管理。设备要求 :规定了生产设备的设计、选型、校验、清洗、维护和使用记录。生产过程控制 :概述了工艺设计和过程单元操作的详细要求,包括配料、粉碎、混合等。物料管理 :强调了物料的接收、储存、分发、退库以及检验与放行的管理。以上仅为部分要点,请阅读原文,深入理解监管要求。