|
首页
>
资讯
>
FDA 定稿局部用药 Q3 指南,划分三个对比层次
出自识林
FDA 定稿局部用药 Q3 指南,划分三个对比层次
2026-03-23
3月16日,FDA定稿《ANDA 提交的局部用药的物理化学和结构(Q3)表征》指南,与2022年征求意见稿相比并无显著改动。该指南旨在为仿制药申请人提供关于皮肤用(包括皮肤及黏膜)液体和/或其他半固体制剂的物理化学及结构(Q3)特性研究的建议。
该指南的核心在于指导申请人如何开展Q3表征以实现两个主要目标:第一,识别拟申请的仿制局部用药的剂型;第二,描述药品中可能对其性能至关重要的属性,以支持生物等效性(BE)的论证。
Q3表征的10个维度
该指南详细阐述了构成局部用药全面Q3表征的10类典型研究内容:
这一框架与我国CDE于2026年1月23日发布的同类指南《局部起效化学仿制药物理化学及结构(Q3)特性研究技术指导原则(征求意见稿)》中明确的10项 Q3 特性研究内容基本一致。
指南建议,Q3表征研究内容应在eCTD的“3.2.P.2 产品开发”提交。同时,该部分也应包含关于影响仿制药Q3属性的因素(如与生产工艺相关)的信息。为确保数据的代表性,指南要求比较Q3时应使用至少三批仿制药和三批(如可获得)参比标准品(reference standard)进行。
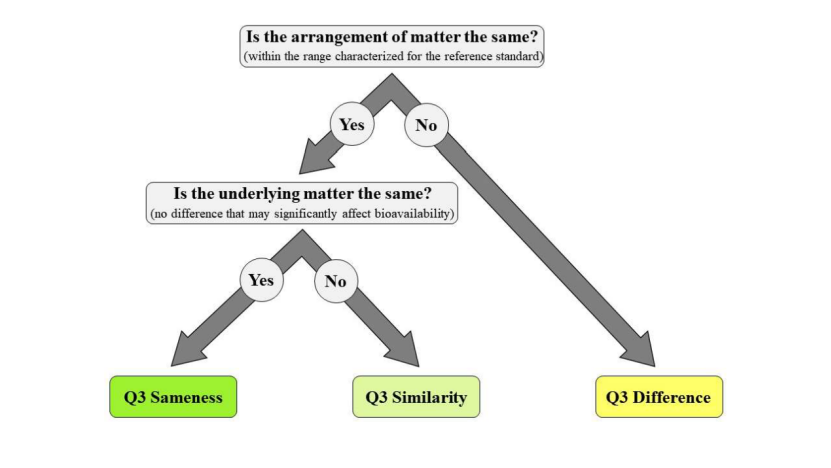
FDA划定Q3可比性的三个层次
除了研究框架,FDA还定义了仿制药与参比制剂对比Q3属性时的三个层次:“相同”(sameness)、“相似”(similarity)和“差异”(difference),并阐述了它们各自的潜在意义,尤其是对BE的支持力度。
Q3相同
仿制药需满足:
- a. 仿制药多批次表征的Q3属性均满足以下条件之一:1)其处于参比标准品(可能经多批次表征)对应Q3属性范围内,或2)由FDA判定处于参比标准品的可接受变异范围内。
- b. 仿制药与参比标准品在组分或构成上不存在可能显著影响全身或局部利用度的差异。
指南认为,证明仿制药与其参比标准品Q3相同可大幅降低BE潜在失败模式的风险。此外,Q3相同的皮肤用溶液剂型通常可豁免体内BA或 BE。
Q3相似
仿制药需满足:
- a. (同 Q3 相同标准a)
- b. 仿制药与参比标准品在组分或构成上,存在可能显著影响全身或局部利用度的差异。
指南认为,证明Q3相似同样可大幅降低BE的许多潜在失败模式风险,但无法排除因仿制药与其参比标准品在组分或构成上的特定差异所导致的风险。因此,Q3相似的皮肤用溶液剂型不能豁免体内BA或BE。
Q3差异
仿制药需满足:
- a. 仿制药经多批次表征的一项或多项Q3属性既未处于参比标准品(可能经多批次表征)对应范围内,也未被FDA判定处于参比标准品的可接受变异范围内。
- b. 仿制药与参比标准品在组分或构成上,可能存在影响全身或局部利用度的差异。
指南指出,鉴于导致Q3差异的原因多样,对于申请人还可提供何种额外证据以支持BE论证,指南无法给出具体建议。
最后,FDA 明确不会向仿制药申请人传达关于其仿制药是否与参比标准品Q3相同或相似的结论,但FDA打算就申请人提交的处方是否适合用于BE进行沟通。
FDA此举应是出于对商业机密的保护。此前在仿制药Q1/Q2(定性/定量)评价方面FDA也不得不如此处置,但最近的立法授权FDA公开参比制剂具体处方组成用以支持Q1/Q2研究。
识林-实木
识林®版权所有,未经许可不得转载
【文件概要】
该文件针对软膏剂、乳膏剂、凝胶剂等皮肤外用或经皮给药的局部起效化学仿制药,提出物理化学及结构(Q3)特性研究的技术要求。指南涵盖10项核心研究内容:性状(颜色、质地等)、相态(显微成像表征)、微粒结构(粒度分布、乳化类型)、原料药晶型(混悬制剂)、密度(生产影响)、酸碱度(含水制剂缓冲体系)、油相基质(高含量组分分析)、流变特性(非牛顿流体黏弹性及触变性研究)、水分活度/干燥速率(挥发性成分评估)及形变相关特性(包装系统影响)。研究需基于参比制剂对比,采用已验证方法,确保数据科学性和代表性。差异分析需证明不影响质量及安全性。文件整合FDA、EMA及药典等国际标准,为仿制药一致性评价提供技术依据。 【适用范围】
本文适用于中国境内研发的局部起效化学仿制药(软膏剂、乳膏剂、凝胶剂),针对皮肤外用或经皮给药途径。适用企业包括仿制药研发企业、CRO/CDMO及质量控制部门,不涉及创新药、生物制品或口服制剂。 【影响评估】
本文细化Q3特性研究标准,提升仿制药质量对比的严谨性,可能增加研发阶段理化表征的测试成本和时间。企业需调整药学研发流程,强化方法学验证和数据论证,以满足监管对仿制药“质量一致性”的更高要求。 【实施建议】 - 必读岗位:
- 研发(药学): 按指南10项内容设计实验,重点优化流变特性、微粒结构等关键参数。
- QA/QC: 审核方法学验证数据,确保检测覆盖全部Q3特性指标。
- 注册: 在申报资料中整合对比研究数据,差异部分需附风险评估依据。
以上仅为部分要点,请阅读原文,深入理解监管要求。 适用岗位: - QA(质量保证)
- 注册(药品注册)
- 研发(药品研发)
- 临床(临床研究)
工作建议: - QA:确保所有Q3表征测试符合FDA指南要求,并监控测试结果以保证产品质量。
- 注册:在提交ANDA时,包含必要的Q3表征数据以证明仿制药与参比制剂的等效性。
- 研发:进行Q3表征测试,以全面了解产品特性,并支持BE研究。
- 临床:在设计临床试验时,考虑Q3表征结果对生物等效性的影响。
适用范围:
本文适用于提交ANDA的局部药品,包括液体基和/或其他半固体产品,如软膏、乳液、凝胶等,适用于美国市场。 要点总结: - Q3表征目的:明确了Q3表征用于识别仿制药的剂型和描述可能影响产品性能的关键属性。
- Q3表征类型:区分了基本Q3表征(描述剂型)和全面Q3表征(详细描述关键属性)。
- Q3比较:强调了比较仿制药与参比制剂Q3属性的重要性,以支持生物等效性(BE)的证明。
- Q3属性的比较结果:定义了Q3相同性、Q3相似性和Q3差异性的概念,并讨论了它们对BE证明的影响。
- 与FDA的沟通:鼓励ANDA申请者在开发过程中与FDA沟通,以确保Q3表征方法和BE策略的适宜性。
以上仅为部分要点,请阅读原文,深入理解监管要求。 【文件概要】
该指南针对通过简化新药申请(ANDA)提交的局部用药品(包括液体和半固体剂型,如软膏、乳膏、洗剂、凝胶等,但不包括局部溶液剂)的理化与结构(Q3)表征提供建议。指南提出通过基础Q3表征(包括外观与质地、相态、物质结构组织)确认剂型,并通过综合Q3表征(涵盖10类属性,如流变行为、多晶型、pH等)建立产品关键性能的详细档案,以支持生物等效性(BE)证明。指南定义了Q3属性“相同”“相似”及“差异”的判定标准:若测试产品与参比制剂的Q3属性相同,可显著降低BE失败风险,可能豁免体内BE研究;若属性相似,需额外证据解决成分差异对BE的影响;若存在差异,则需进一步评估潜在风险。指南建议申请人结合产品特异性指南(PSG)设计Q3测试,并鼓励通过预ANDA会议或受控函与FDA沟通开发策略。 【适用范围】
本文适用于美国FDA监管下通过ANDA途径申报的局部用仿制药(非溶液剂型),包括软膏、乳膏、凝胶等半固体或液体剂型,涉及化学药及部分生物制品。主要面向仿制药企业(包括大型药企、Biotech及CRO/CDMO),不适用于创新药或原料药开发。 【影响评估】
本文要求仿制药企业对局部用制剂进行更全面的Q3表征,增加了研发阶段的分析成本和时间,但通过明确BE证据要求,可减少后期监管风险。对Q3属性“相同”的产品可能简化BE流程,而存在差异时需额外研究,可能延长审批周期。企业需调整质量控制策略以符合新指南要求。 【实施建议】 - 注册(RA):必读。需将Q3表征数据整合至ANDA的3.2.P.2章节,确保符合FDA格式要求;提前与FDA沟通BE策略。
- 研发(R&D):必读。设计基础与综合Q3测试方案,重点评估剂型一致性和关键性能属性;参考PSG选择表征方法。
- 质量分析(QC/QA):必读。建立多批次Q3属性检测流程,确保数据覆盖低、中、高剪切条件等关键参数;验证分析方法。
- CMC:必读。评估生产工艺对Q3属性的影响,确保与参比制剂的一致性;记录成分差异的合理性证据。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |