首页
>
资讯
>
FDA 罕见病工作:超半数新药批准用于治疗罕见病
出自识林
2024-08-20
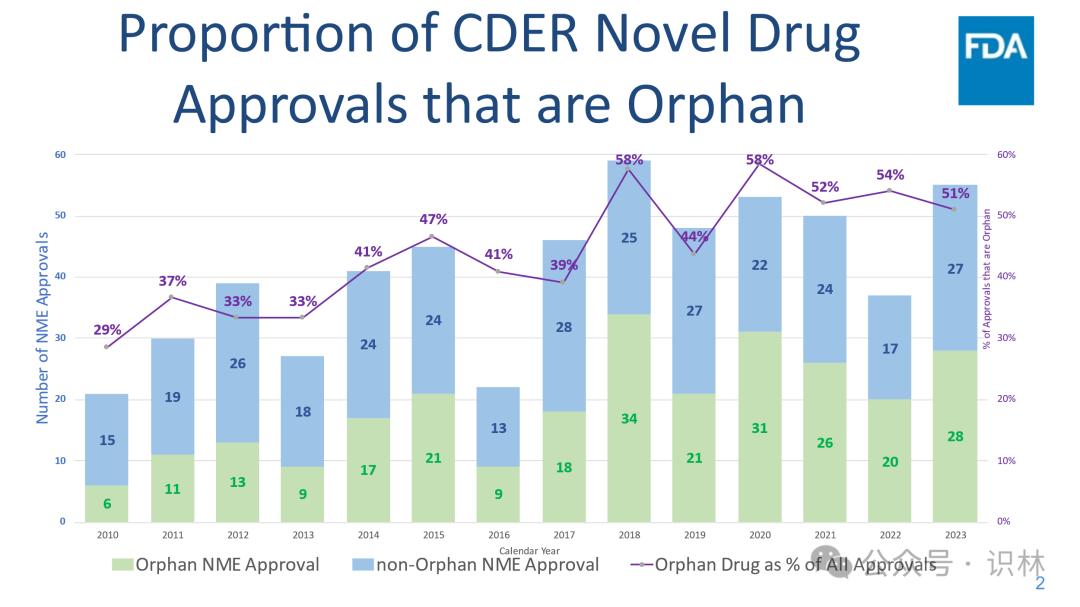
过去四年中,美国批准的孤儿药 数量占到了创新药批准总数的一半以上(见下文图)。孤儿药市场对制药行业具有特别的吸引力,原因有几点:首先,许多公司已经建立了针对这些产品开发的商业模式。其次,这些产品通常有资格获得长达七年的孤儿药专营权(Orphan Drug Exclusivity,ODE),该权利可以在批准之日起七年内提供竞争保护。相较之下,不符合 ODE 资格的新化学实体(New Molecular Entities,NME)首次获批时只能获得五年的 NCE 专营权。此外,由于罕见和极罕见疾病的患者数量少,因此许多此类药物的定价非常高,这不仅使企业能够收回其在药物开发 上的投资,还确保企业能够获得合理的高利润,这种利润模式支持企业继续投入研究其它罕见病的治疗方法。
美国 FDA 药品审评与研究中心(CDER)最近在其网站上发布了一篇题为“
CDER 在罕见病领域工作需要了解的 9 件事 ”的新文章,提供了一些关于罕见病药物的补充观察。下面我们一起来看看。
《孤儿药法案》将罕见病定义为在美国影响不到 20 万人的任何疾病或病症。大约有 2500 万到 3000 万美国人患有一种罕见病(十个人中就有一个患有罕见病)。许多罕见病会危及生命,并且大多数都没有治疗方法。加速安全有效药物的开发是 CDER 的核心使命,CDER 明白在证明治疗罕见疾病的药物的安全性和有效性方面存在独特的挑战。
以下是关于CDER为加速开发治疗罕见病的安全有效药物所做的一些工作:
1. 出于多种原因,针对大约 1 万多种罕见病的药物开发可能很复杂。
由于患者人群少且多样化通常限制试验设计的选择,自然史通常不太清楚,以及缺乏药物开发工具等问题,罕见病药物开发具有独特的挑战性。因此,传统且成熟的临床试验设计可能不支持罕见病的临床研究。由于这些以及许多其它原因,许多罕见病只有很少或根本没有治疗方法可供患有这些疾病的患者使用。
2. 2022年,CDER启动了加速罕见病治愈(ARC)计划,以帮助弥合罕见病药物开发的复杂性与患者迫切需求之间的差距。
CDER 的 ARC 计划通过促进创新的科学设计,加深对监管政策的理解,以及与患者及其倡导者、学术研究人员、临床医生和药物研发 人员的互动,努力增加罕见病的治疗数量。ARC 计划是由 CDER 罕见病团队(RDT)管理的整个 CDER 范围的计划,建立在 CDER 中心主任办公室、新药办公室、转化科学办公室和沟通交流办公室之间的合作之上。对其使命至关重要的是,ARC 计划还汇集了整个机构的办公室和中心,例如局长办公室,包括儿科治疗办公室和孤儿产品开发办公室,生物制品审评与研究中心(CBER),器械和放射健康中心(CDRH),以及肿瘤学卓越中心(OCE)。自启动以来,ARC 计划已成为赋予罕见病社区权力以利用其集体经验和专业知识来推动进展的渠道。
3. CDER 和 CBER 正在合作建立 FDA 的罕见病创新中心。
FDA的罕见病创新枢纽(Rare Disease Innovation Hub) 将针对所有罕见病,但将特别关注针对较小人群或自然史多变且尚未完全了解的疾病的产品。它将利用 CDER ARC 计划的活动来加强现有的跨中心合作。
该枢纽将具有三个主要功能:
在与 CDER 和 CBER 交叉的问题上,作为罕见病社区(包括患者和护理人员团体、贸易组织以及科学/学术组织)的单点联系和参与。该枢纽将帮助更大的罕见病社区了解 FDA 内影响罕见病患者的重要交叉点,例如医疗器械 (包括诊断测试)和组合产品 。
加强中心间协作,解决与罕见病产品开发相关的常见科学、临床和政策问题,包括与产品审评相关的跨学科方法,并促进办公室和中心之间的一致性。
4. 有四个加快审评 计划可帮助尽快提供严重疾病未竟需求的药物,其中许多是罕见病。从 2015 年到 2023 年,CDER 中 88% 的新药和生物制品批准使用了至少一个加快计划。
快速通道 - 快速通道是旨在加快用于治疗严重疾病并满足未竟医疗需求的药物的开发和审评的过程。快速通道认定的好处可能包括与 FDA 更频繁的会议以及“滚动审评”的选择(上市申请的所需部分在不同的时间进入FDA)。
突破性疗法 - 突破性疗法认定是一个旨在加快开发和审评用于治疗严重疾病的药物的过程,在该路径下,初步临床证据可用来证明在具有临床意义的终点上比现有疗法有实质性改进。FDA 就有效的药物开发、涉及高级管理人员的组织承诺以及该计划下的“滚动审评”选项提供了加强的指导。
优先审评
加速审批 - 加速审批允许更早批准治疗严重疾病、相比现有疗法提供有意义的优势,并证明对替代终点 有影响的药物(一种被认为可以预测临床获益的衡量标准,但其本身不是临床获益的衡量标准)。申办人必须在批准后临床研究 或确证性试验 中核实和确认临床获益。
5. FDA 继续看到批准用于治疗罕见病症或疾病的药物的数量和百分比呈上升趋势。
“创新药”是指从未在美国批准或上市的新药。出于FDA审查的目的,药物被归类NME。
2023 年,超过一半(55 个中的 28 个,或 51%)CDER 批准的新药被批准用于预防、诊断或治疗罕见疾病或病症。其中包括:
弗里德赖希共济失调的首个治疗方法,这是一种损害神经系统的遗传性退行性疾病。
活化磷酸肌醇3-激酶δ(一种损害免疫系统的遗传性疾病)的首个治疗方法。
6. CDER 最近成立了一个新的专家委员会,以帮助 FDA 探索与遗传代谢疾病药物开发 相关的复杂问题。
2023 年,CDER宣布成立新的遗传代谢疾病专家委员会(Genetic Metabolic Diseases Advisory Committee,GeMDAC) 。在 CDER 罕见病和医学遗传学处的权限范围内,遗传代谢疾病是干扰人新陈代谢的罕见疾病,新陈代谢是人体将食物转化为能量并清除废物和不健康物质的能力。GeMDAC 将就在研或已提交上市许可 的用于治疗遗传代谢疾病的人用药和生物制品 的安全性和有效性向 FDA 提供建议。
7. 在 ARC 计划下,CDER 制定了促进和授权罕见病药物 开发人员的学习和教育(Learning and Education to Advance and Empower Rare Disease Drug Developers,LEADER 3D)计划,以更好地了解将罕见病产品推向市场的障碍。
作为该计划的一部分,RDT 与设计和实施罕见病临床试验的利益相关者进行了接触。目的是确定可以从教育材料的开发或扩展中受益的监管主题,并为持续的教育工作和外联提供建议。通过此次参与,包括对罕见病药物研发界的重点访谈和一份公开卷宗,CDER 发布了 LEADER 3D 外部利益相关者分析公开报告。
基于 LEADER 3D 公开报告的调查结果,RDT 正在开发教育资源,以应对将安全有效的药品和生物制品推向市场以供罕见病患者使用的挑战。
8. CDER 与其它 FDA 中心和办公室合作,以帮助推进罕见病药物的开发,例如支持临床试验推进罕见病治疗(Support for clinical Trials Advancing Rare disease Therapeutics,START)试点计划和罕见病终点推进(Rare Disease Endpoint Advancement,RDEA)试点计划。
CDER 和 CBER 发起了 START 试点计划,希望通过该试点获得的见解将提供关于如何最好地促进更有效地开发可能挽救生命的罕见病疗法的信息,并将帮助申办人产生高质量、可靠的数据以支持未来的新药或生物制品许可申请 。选定的参与者将经常获得 FDA 工作人员的建议,以解决特定于产品的开发问题,包括临床研究设计、对照组的选择以及对患者群体选择的微调。这些增加的互动包括解决可能延迟或阻止有前景的新产品进入关键临床试验阶段的早期开发问题,以及确保对促进产品开发所需的信息和数据的清晰理解。
RDEA 试点计划是 CDER 和 CBER 的一项联合倡议,支持疗效终点(或结局)的开发,以及治疗罕见病(包括儿童罕见病)的药物和生物制品的及时批准。该计划专为正在申请研究用新药(IND )申请或罕见病处于 IND 前阶段的药物研发人员而设计。该计划也适用于尚未制定有效的药物开发 计划但已经或正在开始自然史研究以检查拟议终点的申办人。
9. CDER 关键路径研究所(C-Path)合作在 ARC 计划下开展了几个罕见病项目。这些公私合作伙伴关系将各种罕见病利益相关者(如倡导团体、学术界、药物研发人员、患者和其他合作伙伴)与 FDA 联系起来,以帮助确定罕见病药物 开发挑战的解决方案。
“溶酶体疾病联盟”旨在连接 FDA、领先的学术机构、药物研发人员、患者团体和非政府组织,以解决溶酶体疾病(由于酶缺乏而导致各种有毒物质在身体细胞中异常积聚为特征的遗传性代谢疾病)患者未满足的药物开发需求。
“罕见神经退行性疾病的关键路径”旨在促进对神经退行性疾病的理解,同时促进肌萎缩侧索硬化症(ALS)等罕见神经退行性疾病的治疗方法的开发。
编译:识林-椒
识林® 版权所有,未经许可不得转载。