首页
>
资讯
>
PMDA 发文展示多项举措促进创新药进入日本
出自识林
PMDA 发文展示多项举措促进创新药进入日本
2025-09-03
近日,日本药品和医疗器械管理局(PMDA)在Clinical Pharmacology & Therapeutics上发表文章,系统性介绍一系列举措,以应对日本当前面临的“药品滞后(Drug Loss)”问题,即海外已获批新药在日本未能及时申报或上市的现象。日本虽被我国赶超,但仍位列全球第三大药品消费市场,我国新药近年来也频频在日本上市 。以下内容供旨在走向全球的创新药企参考。
文章指出,自2004年成立以来,PMDA已显著缩短新药审评时间。2005年中位总审评时间超过600天,至2023年已降至333天(按CIRS报告 ,2024年进一步缩减至290天),已达到与FDA等国际监管机构相当的水平。然而,日本在欧美地区多中心临床试验 (MRCT)中的参与率仅约57%,在美国获批的新分子实体(NME) 中有65%未在日本上市。
PMDA认为,一个主要原因是药物研发 主体的变化。近年来,新兴生物制药公司(Emerging Bio-pharmaceutical Companies, EBP)逐渐成为创新药研发的主力。据统计,未进入日本的新药中近半数由EBP开发。
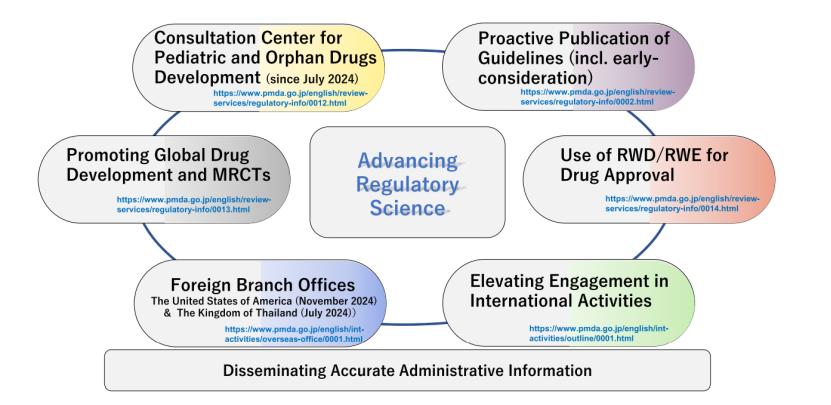
针对这一挑战,PMDA与厚生劳动省(MHLW)协同推出多项措施,其中六项关键措施如下:
设立儿科与罕见病药物研发咨询中心(Consultation Center for Pediatric and Orphan Drug Development, CCPODD),为制药企业与学术机构提供科学建议与法规支持,并对相关咨询实施费用减免。该中心通过发布最新技术指南与监管要求,促进儿科与罕见病药物 的高效开发。
积极发布技术指南与早期考量(Early Consideration)文件。早期考量文件并非正式指南,而是类似欧盟思考性文件(Reflection Paper)的参考文件,旨在为尚未成熟的技术领域提供前瞻性科学观点。2024财年已发布多个早期考量文件,涵盖肿瘤学I期临床试验统计设计、药物-药物相互作用 生物标志物 应用、以及肺动脉高压药物开发等方面。
发布新指南明确在日本开展MRCT前是否需进行I期临床试验 。2024年10月指南进一步说明,罕见病药物在某些情况下可豁免日本临床试验数据提交。PMDA还与指定临床试验机构合作,提高试验效率并降低成本。
推进真实世界数据(RWD)与证据(RWE)支持药物审评,尤其适用于难以实施随机对照试验的罕见病领域。PMDA近期发布关于外部对照(External Control)的早期考量文件,系统阐述RWD/RWE 在研究设计、分析与解释各环节的关键考量。
开设海外办事处。PMDA于2024年7月在泰国曼谷设立首个亚洲办事处 ,并于同年11月在美国华盛顿设立办事处 。亚洲办事处致力于推动与亚洲监管机构的协调与合作,支持包括ARISE(东南亚与东亚学术研究组织联盟)和ATLAS(亚洲癌症临床试验网络)在内的多项区域临床试验 倡议。美国办事处则重点支持EBP了解日本法规并提供开发策略咨询。
PMDA还通过ICH,PIC/S,国际药品监管机构联盟(ICMRA)等多边机制,加强与全球监管机构的协作。其亚洲培训中心(PMDA-ATC)自2016年起已为65个国家超过3100名监管人员提供培训,提升亚洲地区监管能力。
除上述措施外,PMDA的其他措施还包括:(1)日本优先审评路径(Sakigake)和附条件批准等加速审批 途径;(2)近年来,关于工程化病毒载体的《卡塔赫纳议定书》(Cartagena Protocol)实际审查也得到了显著改进;(3)创建了一个英文的网页 ,以帮助外国公司更好地了解日本的法规。
此外PMDA还提及中央伦理审查委员会(Central IRB)的应用,针对创新药物的定价优惠与税收抵免措施,以及2025年2月内阁批准的第三期医疗五年计划明确支持科技创新与药物研发。
文章最后,PMDA表示自己的大门将总是向全球创新药企敞开(doors are always open)。
识林-实木
识林® 版权所有,未经许可不得转载