|
首页
>
资讯
>
【识林社区】优质问答集锦5
出自识林
2022-06-25
以下是近期识林社区中的部分优质问答:
Q1. 首次药厂厂房建设,验证工作如何开展?
完全的一个新药厂建设,首次验证如何开展,必须全都委托第三方验证吗?还是可以先将实验室建设好了,委托第三方将实验室空调系统和必要的实验室设备就开展生产厂房的验证。有没有什么好的实践经验?
社区用户@阳光蒲照:
《药品生产质量管理规范(2010年修订)》第一百三十八条规定:企业应当确定需要进行的确认或验证工作,以证明有关操作的关键要素能够得到有效控制。
首先应当明确,验证主体是药品生产企业本身。在此基础上,才能进一步讨论验证方案由谁制定,由谁进行确认。问题中,相关验证文件由药品生产企业批准,最终均能够保证被确认的对象有效,从理论上而言没有什么不可以。
但是,完全将厂房设施和设备的验证委托给供应商进行有时不能达到验证的效果,因为供方可能是建筑或设备方面的专业人员,但其对产品特性的了解一般不如生产企业人员,而设施设备的验证是必须结合产品特性开展的,其验证结果是为生产产品而服务的。
至于文件是全英文合不合适,药品生产质量管理规范没有进行强制要求。企业应考虑企业的人员素质:首先,企业人员应当能够对这些文件认真进行审核,否则无法进行批准;其次,无论由哪方来完成验证,必须确保本公司相关人员完全读懂这些文件,并掌握验证的内容、过程和结果。
以上内容来源识林新建药厂验证问答
希望能帮到您!
社区用户@吹口琴的猫:
法规要求已经回答了,我想说一下实际操作层面的:
最好进行第三方的委托:
1. 因为新建厂房,仪器设备等都没有进行验证,尤其是实验室仪器没有进行验证,这时是无法开展的,如果一步步等实验室先进性验证后,在开始其他验证没那个会很费时间,虽然省了委托费,但是进度会受到影响。
2. 因为新建厂房,所以不可避免的会有很多新人,第一次就进行验证,会出现大量的问题,缺陷,不如直接委托第三方,其实也有和个培训的过程,对本公司人员进行培训。
3. 即使是委托验证没要必须公司内相关人员深入参与,绝不能当甩手掌柜。我的经验,及时是国内一流的验证服务商,如果公司人不深入参与,效果也不好,可靠性不能保证。
4. 所有文件必须经过本公司人员严格审核,不能依赖于被委托方,因为验证公司的是模板化的,针对性不强。要针对本公司特点。
5. 如果自己验证,要注意先对人员资质进行确认,人员首先符合要求,一定要注意时间逻辑,前置条件。
Q2. 如何快速查询一个药物的全球首次获批时间?
7月23日CDE发布的风险分析与管理计划撰写指导原则(征求意见稿),正文中有一行需要填写该药物的全球首次获批时间,请问大家是如何快速准确的查询到这个结果?
社区用户@南瓜:
1. 收费数据库:
如果贵公司购买了例如Cortellis、Pharmaprojects等收费数据库,那么查询绝大多数药物的全球上市时间就比较容易了,在数据库中直接检索即可,不再赘述。
2. 免费检索途径:
免费检索途径相比收费数据库会更费时且需要一些技巧,有时可能需要同时结合多种途径相互佐证,主要的途径有如下几种:
(1) 搜索引擎:
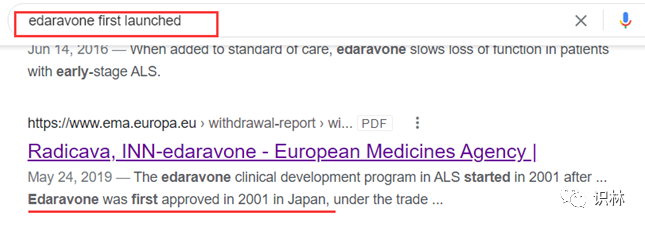
此方法是最直接的方式,推荐使用Google或Bing等搜索引擎,以英文关键词“药物名称+first launched/approved/marketed/developed/introduced”检索,通过搜索出的文章或者文献来获取某药物的最早上市时间。
以依达拉奉为例,在Google中搜索“edaravone first launched”,可以搜索出多篇官方或非官方的文章或文档,都可以确定该药物最早于2001年在日本上市。
此方法适合大多数药物的检索,但是信息的准确度需要根据信息来源是否可靠进行评估或通过下述其它检索方式进一步确认。
(2) 上市公司年报查询
由于上市公司需要向投资者披露公司的信息,因此可以在上市公司年报中查找某药物的批准情况。此方法适用于查询近年来上市公司的一些新药,上市时间比较久远的药物一般不适用。在确定某药物的原研公司后,查询该公司是在哪个国家或地区的证券交易所上市,可选择在监管机构或上市公司官网下载该公司年报文件。
这里只举例比较常见的在美国证券市场上市的情况,以Intercept制药公司的OCALIVA为例:
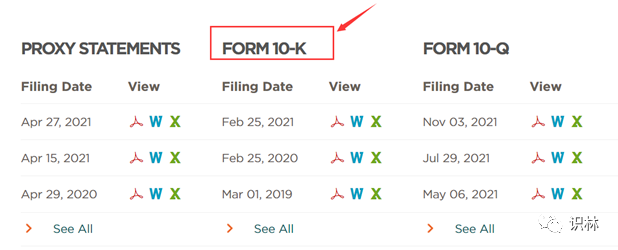
SEC网站下载:不管是在纽约证券交易所 (NYSE) 还是纳斯达克证券交易所 (NASDAQ) 上市的公司,都归美国证券交易委员会 (SEC) 监管,因此可在SEC官网下载年报文件,直接在SEC官网搜索公司名称或股票代码就可以搜索出公司相关披露文件。需要注意的是,注册地在美国的公司年报为10-K文件,季报为10-Q文件,注册地在美国以外的公司年报为20-F文件,选择相应的文件下载即可。
官网下载:除了SEC以外,上市公司一般会在官网的Investor Relations (IR)板块提供年报文件下载。进入Intercept公司官网,点击上方“INVESTORS & MEDIA”进入IR页面,
再点击左方的“FINANCIAL INFORMATION”
进入财务信息页面后即可下载年报10-K文件。
无论是通过SEC还是官网下载Intercept公司的2021年报文件后,可以在其中找到关于OCALIVA的上市情况,可以看到OCALIVA最早是在2016年5月在美国被批准上市,2016年12月被欧盟批准.从2017年1月开始,陆续获得加拿大、以色列和澳大利亚等国批准。
有些时候在最新的年报文件中无法找到某药物的最早上市信息时,可以多往前翻几年的年报也许可以找到相关信息。因为是原研公司自己公布的信息,所以本检索途径相对于其它途径来说是最准确的,但有些药物的最早上市时间可能通过此途径无法获得。
(3) 日本IF文件
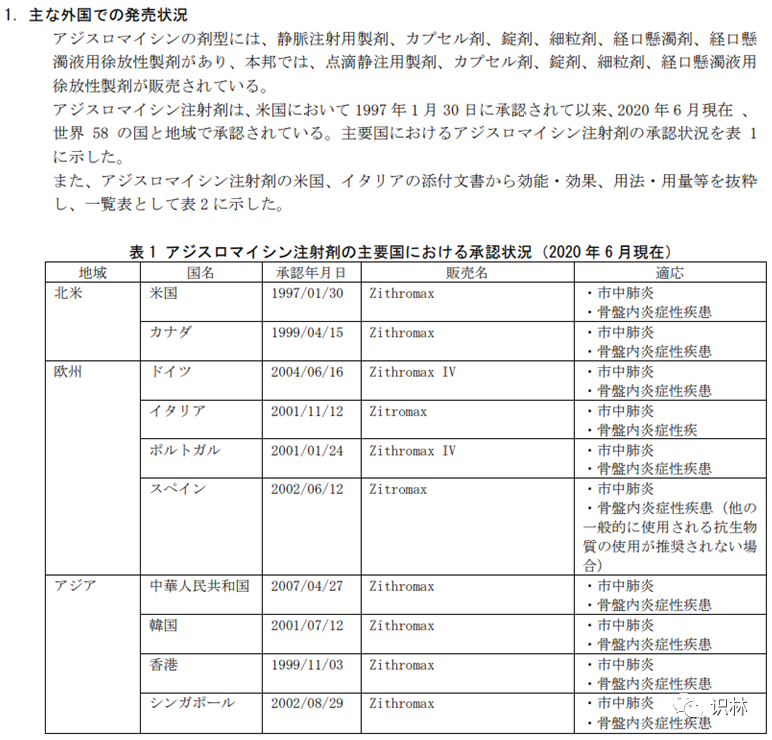
上市相对久远的药物的相关信息总是很难检索,这时我们可以借助日本的IF文件来获得信息。药品研发人员多通过检索日本IF文件来获得在日本上市药品的处方、包材、稳定性等药学信息(IF文件具体下载方法请参考网上相关教程,不再赘述),但其实日本IF文件是由固定格式要求的,其中“開発の経緯”部分可以看到该公司对某药物上市历史情况的综述,而“主な外国での発売状況”部分中则是该药物在日本国外的上市情况,如果是原研的IF文件,那这两个部分内容均较为准确。如果是仿制药的IF文件,则本部分内容可作为参考。
例如,注射用阿奇霉素。化合物最早是由PLIVA公司开发并授权给辉瑞公司进行部分地区的销售,该药物上市时间较早且有多种剂型和规格。打开辉瑞公司注射产品的IF文件,可以看到在“開発の経緯”部分中详述了此药物的开发过程,可以看到辉瑞公司的注射剂最早是于1997年在美国上市。
而在“主な外国での発売状況”我们可以看到各主要国家的上市时间,同样也可以确定最早上市时间。
该方法只适用于在日本由上市的药物,尤其是日本公司参与研发的药物“開発の経緯”部分会撰写的非常详细。但有时原研公司可能只会写出在日本的开发背景,不会写出全球的上市情况,这时需要结合其它方法来进一步确定。
(4) 免费数据库
有一些免费数据库也可以检索药物的上市情况,例如Drugbank等数据库,可参见此页其它回答中的检索方式。但是免费数据库中的数据可能存在不准确或不全面等问题,所以需要同时结合其它方式确定。
(5) 各国药监局查询
此方法是通过对全世界主要国家或地区(中国、美国、欧盟、日本、韩国、俄罗斯、加拿大、阿根廷、巴西、瑞士等)的药品监管部门进行穷举式的检索来确定,此方法较为费时,各国家上市药物查询方式请参考网上相关教程,此处不再赘述。
但是,此方法可以作为以上任一方法的补充或者作进一步确认时使用。
Q3. QA数量与生产人员数量比例多少合适?
现在企业严格控制各部门增加人员,尤其是QA部门。
想了解下,对于药企,QA跟生产人员的比例多少算合适?
社区用户@牧魂:
说一下对这个问题的看法,看似简单的一个问题,实则反应出现阶段药企的现状,使得我们的管理人员,有些不知所措,奈何企业老板更难,毕竟在外部环境下,所感受到的压力,估计不是普通员工能够感受到的。
1. 这看似是一个法规问题,实则是一个管理问题。尽管说,相关法规有明确规定,需要配备一定数量的专业人员,比如QC,QA等等。但是国内外监管方不会给出指南的,毕竟这是一个管理问题。管理,管理,就是通过管理,来实现利益的最大化。企业生存不易,降本增效永远是企业重点考虑的之一,也是管理人员绩效考核一个较为重要的指标。
2. QA跟生产人员的比例,这两者没有必然联系。因此说,试图推导出一个合适的比例,说得严重一些是本末倒置。反推一下,是不是一个合理的比例,就可以实现企业的目标,或者至少符合或者满足现在的需求,更或者符合GMP要求?正反推理,都是没有太多相关性。
3. 实际上老张想了解行业的平均水平,来说试图说服老板或者HR(HR实则也是听从老板的安排或者命令)来降低工作压力,而这个压力通常是来自下属或者生产车间的反馈等。那么即便通过较为科学的统计分析,得出了一个比例,比如8%,那么你去拿这个数据是和老板谈,老板会反问老张一堆问题,不行你试试?当然显然试过,或者不敢,毕竟没有数据。
4. 实际上我们要的不是比例这个无用数据,而是能够支撑你们增加QA人员的一个支持性数据。更有可能只是一些工作没有人去落实,或者一些问题,现有的QA解决不掉或者解决不好造成的压力,而已。
5. 那么实际上,如果我们能够很好的分析现状,类似于差距分析。差距分析的一个基本前提是缕清现状,说白了QA目前需要做哪些工作,并对他们这些工作进行分类。而分类是进行合并,组合,拆分,删除,转移,覆盖,优化,调整等一系列工作,另外可以通过采用一些高效的管理工具和方法来提升效能。现实工作中发现,有些工作并不是需要的,有些工作换一个人干更高效,有些事情可以从纸质版变成电子版等等。
6. 可能说老张这些事情多做了很多,依然觉得人不够,毕竟人不是机器,即便是机器,也是要大修维保的。那么实际上人的潜力,还可以挖掘一下的。还有,QA工作分工越细,实际事情可能几何倍数的增加。还有流程,有的时候完全可以更偏平化,有些不相关的部门,不相关的会议,培训,检查,文件修订,记录版本变更,验证,计量校准,取样工作都是可以变换方法的。
7. 如果还是不行,你把上述这些情况,有理有据的和老板谈,难道老板如此不讲道理?或者老板难道对重要品种,重要车间都不考虑?如果依旧不考虑,你已经是一个管理大咖了,为什么非得这个平台呢?
Q4. 工艺变更后可比性研究的稳定性趋势对比应该怎么做?
场地变更需要进行可比性研究,需要开展稳定性趋势的对比。请问稳定性趋势的对比方法有哪些?
社区用户@吹口琴的猫:
这个问题有两个方面,
1. 一个是可比性研究的判断,这个已经有了现有的回答,就不在重复
2. 是稳定性的趋势如何判断?这个主要看趋势是否正常,也就是有没有OOT,方法有几种:
经验方法:目前在药品稳定性试验中广泛使用的用于确认OOT结果的方法包括:(1)连续三个结果超出某一限度;(2)连续结果之间的差异超过了先前结果与质量标准差异的一半;(3)结果超出了最初结果的±5%;(4)结果超出了之前结果的±3%;(5)结果超出了之前所有结果平均值的±5%⑶。
统计方法:回归控制图法,要求有一定的统计学基础,实施计算比较复杂:如果稳定性数据是随着时间降低的,则平均变化值是负值,警戒限应该包括最小值,如果稳定性数据随时间变化是稳定的,则其平均变化值近似为0,警戒限应该包括上下限,分析警戒是监测实验室错误的,所以建议设定上下限如,OOT警戒限可以用下述公式来规定:
变化值平均 ± (K x 变化值标准偏差)
公式中,K是变异系数,一般取K=3(假设数据是正态分布的)或者取t统计量(f,1-α),f代表自由度,α表示显著性水平。上下限度可以取差异分布的α和1-α的的百分数。这中方法适用于稳定性数据不呈正态分布的情况。
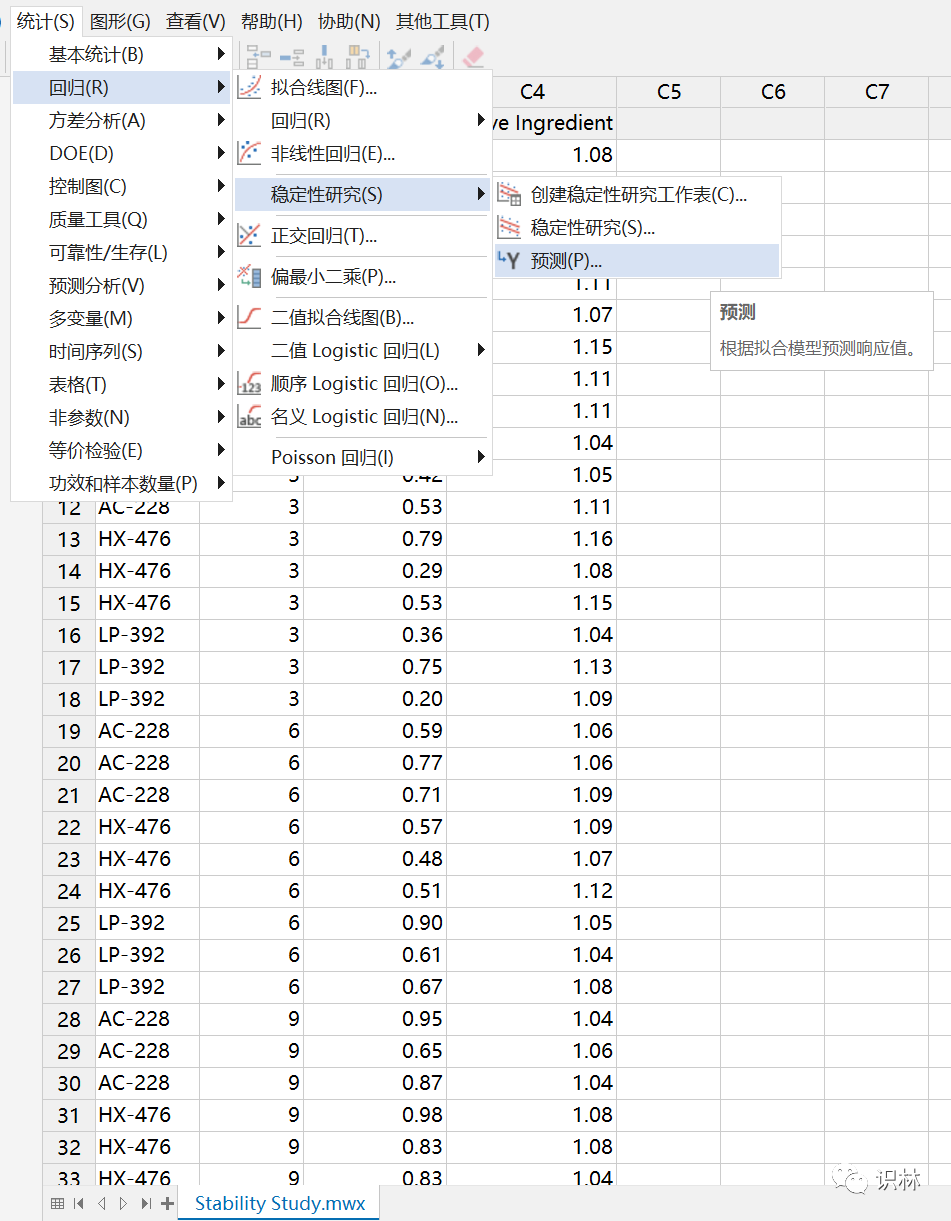
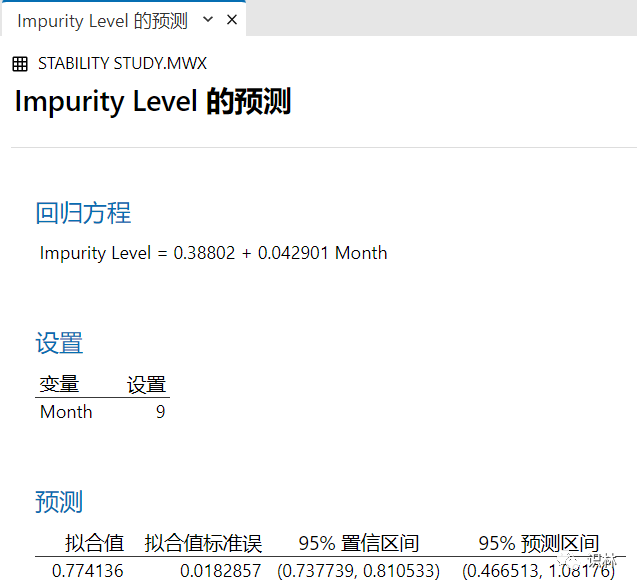
3. MINITAB稳定性的预测功能:(我个人推荐)
最大的差别不能超过预测区间,一般不超过置信区间为好,超过置信区间就可以进行调查。
(登录识林社区,可查看回答中涉及的法规原文链接)
识林社区是一个面向制药业从业者的互动交流平台,主要功能包括问答、文章、课程。
在这里,成百上千的识林用户提出问题,而回答问题的有其他用户,还有识林向导,识林内部团队和专家。
药品生命周期,是大量法规、知识、实践的结合,大家每天都会遇到大大小小的问题,但并不总能得到好的答案。
一个好的答案,不能是一句道听途说,也不仅是从“经验”出发的一句简单判断。更何况,越重要、越复杂的问题,往往并无完全正确、拿来即用的答案。
所以,优质解答,应兼具“依据”、“思考”和“建议”。读完之后,不仅可基于“建议”有所行动,也能审视答者的“思考”逻辑,还可以确认“依据”,并发散学习,真正把答案转化为自己的知识,应对未来其他问题,“鱼”与“渔”兼得。
欢迎用户登录识林社区url,参与问答与讨论。
识林®版权所有,未经许可不得转载
适用岗位解读: - QA(质量保证):必读。应确保所有生产活动符合GMP要求,监控生产过程,保证产品质量。
- 注册:必读。需熟悉GMP规范,以确保注册文件和申报材料的合规性。
- 生产:必读。应根据GMP要求组织生产活动,确保生产过程的规范性。
- 研发:参考。在产品开发阶段考虑GMP要求,为后续生产打下基础。
- 临床:参考。了解GMP对临床试验用药品的特殊要求,确保临床试验的合规性。
适用范围说明:
本文适用于在中国境内从事化学药品、生物制品、疫苗、中药等各类药品生产的企业,包括原料药、制剂等不同注册分类,以及Biotech、大型药企、跨国药企、CRO和CDMO等不同企业类别。 文件要点总结: - 原料血浆管理:强调原料血浆的质量和来源合法性,以及生产过程中病毒去除和/或灭活的严格控制。
- 人员资质要求:明确企业负责人、生产管理负责人、质量管理负责人和质量受权人的资质和经验要求。
- 生产设施和设备:规定血液制品生产厂房、实验室的独立性和专用性,以及防止交叉污染的措施。
- 原料血浆检验与追溯:要求企业对原料血浆进行严格检验,建立追溯系统,确保可追溯性。
- 生产和质量控制:强调生产过程中的温度验证、体外诊断试剂管理、病毒污染和安全风险评估,以及信息化记录的重要性。
以上仅为部分要点,请阅读原文,深入理解监管要求。 岗位必读建议: - QA:负责确保实验室操作符合质量控制要求,监督取样、留样、检验等流程。
- 研发:在设计质量标准和分析方法时,需遵循本文规定。
- 生产:在取样和留样过程中,应遵守本文的详细规定以保证产品质量。
文件适用范围:
本文适用于化学药、生物制品、疫苗和中药等药品类型,包括原料药、中间产品、待包装产品和成品。适用于创新药、仿制药、生物类似药等注册分类。适用于中国药企,包括Biotech、大型药企、跨国药企、CRO和CDMO等企业类别。 文件要点总结: - 实验室职责与布局:明确了质量控制实验室的职责、布局原则和要求,以及人员的组织架构和资质要求。
- 取样与留样管理:规定了取样过程的控制和留样的定义、量、储存要求及记录。
- 物料和产品检验:强调了检验要求,包括待检样品核对、检验、记录和报告书的编制。
- 委托检验管理:阐述了委托检验的原则、应用范围、职责和工作流程。
- 质量标准建立:详细说明了质量标准的设计与制定、审核与批准流程。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |