首页
>
资讯
>
【识林社区】优质问答集锦7:设备验证,仓储,方法转移,天平校验,数字修约,pre-IND
出自识林
【识林社区】优质问答集锦7:设备验证,仓储,方法转移,天平校验,数字修约,pre-IND
2022-11-19
识林社区,目前向全部识林用户免费开放。
大家登陆注册后,就可提问与回答。识林社区详细介绍请见文末
识林邀请了许多来自企业一线的专业人员,作为识林的“向导”,回答大家的疑问。
以下是近期来自向导的优质回答,供大家参考探讨。
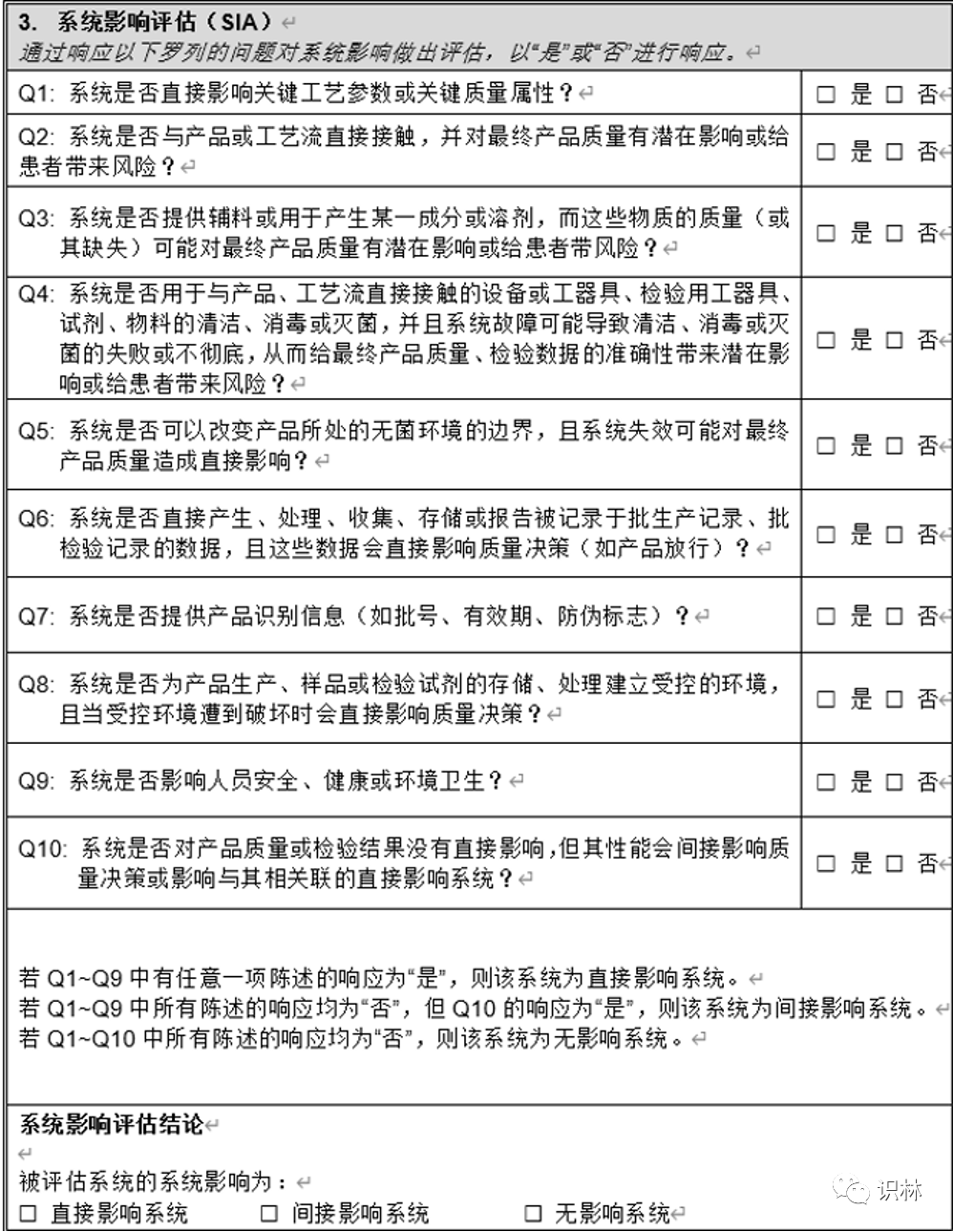
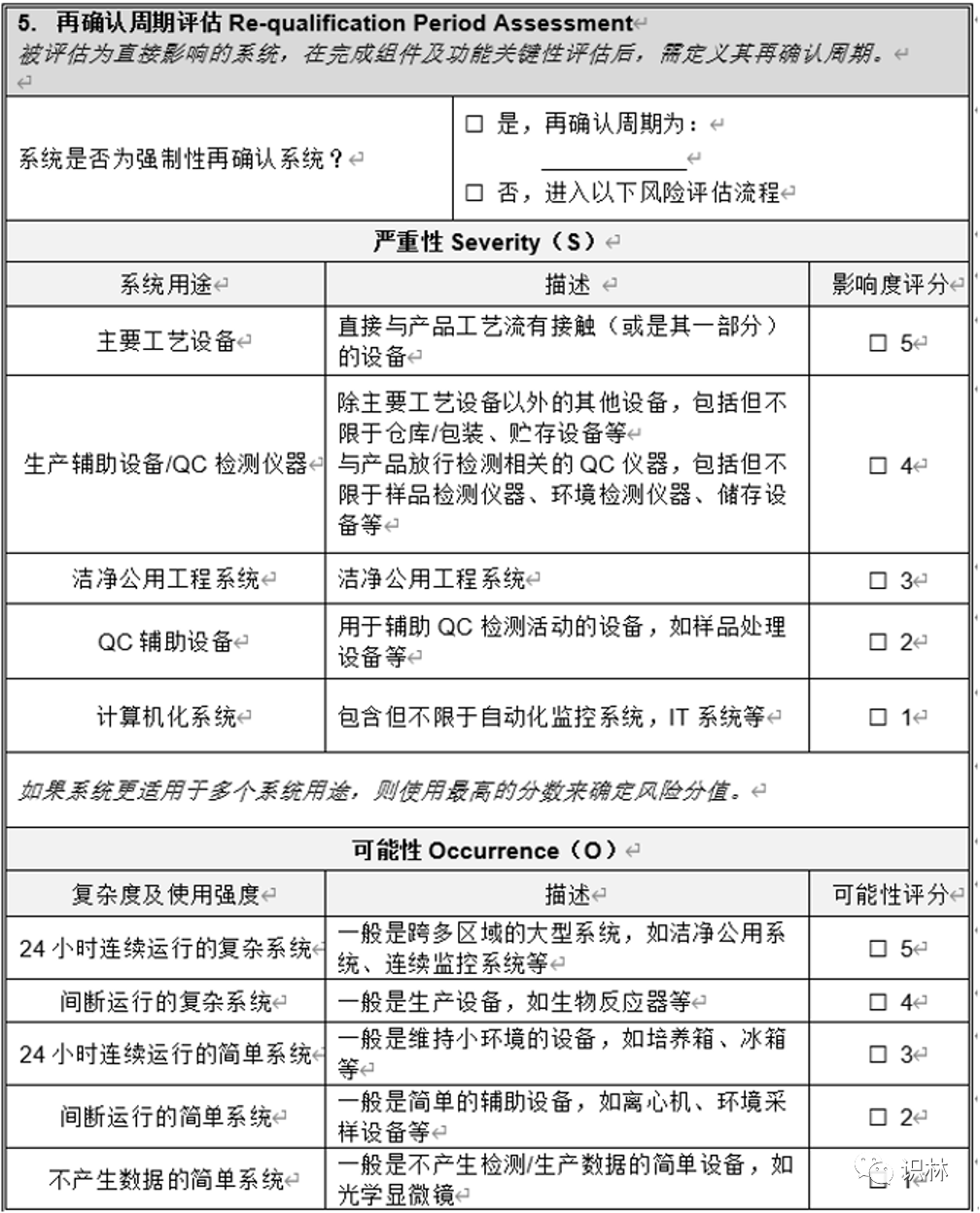
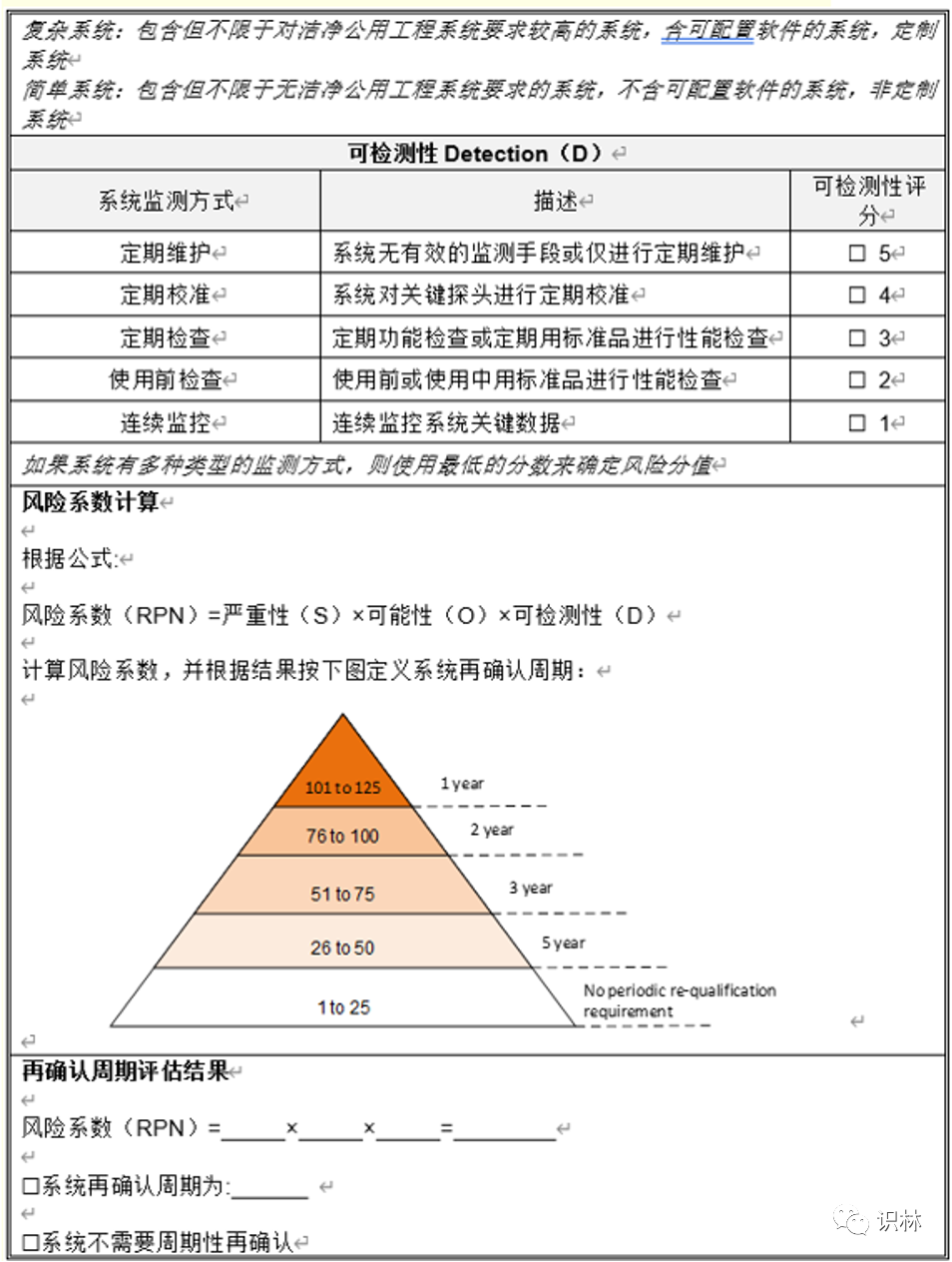
Q1. 如何开展设备、设施的定期审核评估?
— 关于非法规/指南等强制的再验证 周期设备 设施 ,如何开展定期审核评估和周期的确定?
向导@圣人有点冷:
需对设备的关键性进行评估,可参考ISPE调试与确认(第二版)系统影响评估(8个问题),对系统进行分级,分为之间影响系统、间接影响系统、无影响系统,评估为直接影响系统的再对其进行再确认 周期的判定,分为强制再确认周期,这个是法规规定的,如灭菌器,A级洁净区 空调等;非强制性,根据系统的风险,从严重性、可能性、可探测性进行打分并计算RPN。根据RPN确认再确认周期。
同一台设备根据其使用的用途可以制定不同确认周期,例如冰箱,在仓库、生产和QC实验室 储存的物料 是不一样的,自然风险也不一样。
评估可参考下表:
Q2. 原料药遮光储存可以用双层透明薄膜袋加纸板桶包装吗?
— 原料药 遮光储存可以用双层透明薄膜袋加纸板桶包装吗?还是必须用黑色袋子包装再用纸板桶?
向导@浮云之树:
如何选择其实应该从API自身的性质去分析评估。
遮光系指用不透光的容器 包装,例如棕色容器或黑色包装材料 包裹的无色透明、半透明容器;
避光系指避免日光直射;
可以看出,遮光的要求是更严格的,而需要遮光的API肯定是对光更加敏感的,否则完全可以把储存条件改为避光。
其次,对遮光的定义也写了,可以是黑色材料包裹的无色透明容器,因为黑色材料已经可以完全隔绝光线了。并非强求内容器一定是黑色袋子
所以你们应该考虑三点:
1. 该API 的光稳定性 如何,是否真的需要遮光。
2. 如果确要遮光。储存容器(外)要满足不透光的要求,双层塑料袋当然可以,但是你们的纸板桶能做到黑色袋子那样的隔绝效果吗?如果能证明同等效果就可以用。不要主观的认为纸板桶一定可以,要有证据。很多纸板桶是有一定的透光的。
3. 建议还要考虑到在制剂投产中会不会存在,把塑料袋装的API取出桶后还会放置一段时间的情况,如果有那么最好还是要把内容器换为双层塑料袋再外套黑色袋。
当然,不确定你说的黑色袋子是不是就是直接用黑色袋作为内容器。考虑到可能会有溶出什么的,建议还是以透明薄膜袋作为内容器。
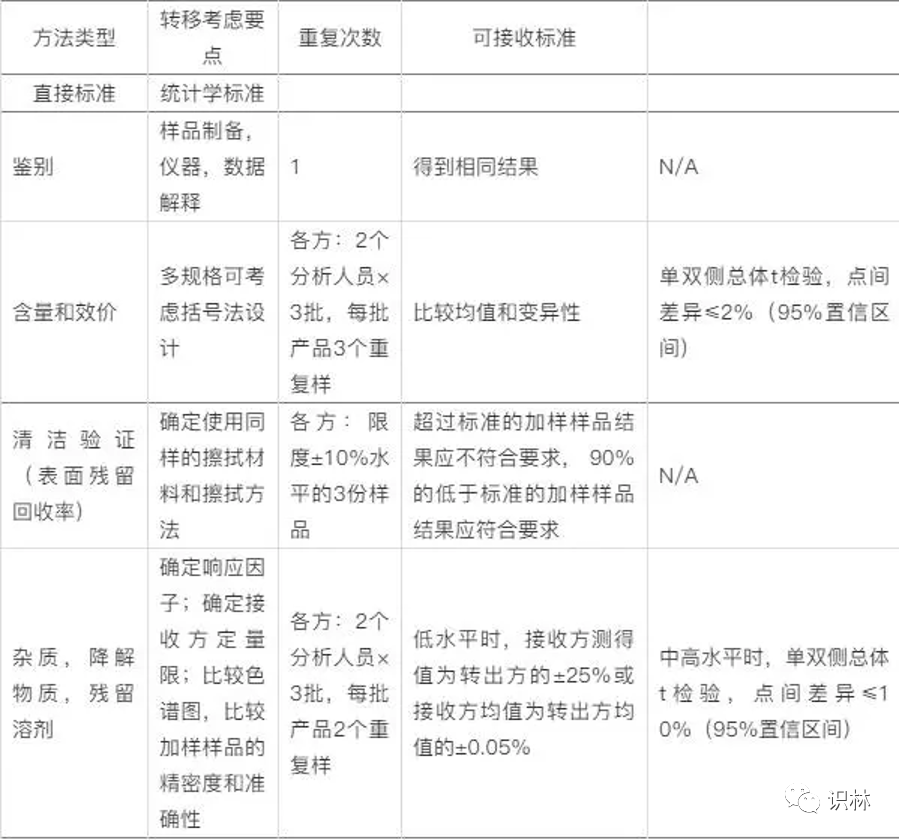
Q3. 方法转移对比实验的可接受标准?
向导@Yiran
2022.11.07-08 中检院注册检验 培训课程中“分析方法 的开发及转移 ”PPT中给出的WHO建议的对比检验 可接受标准 如下表,供参考,希望对你有帮助!
Q4. 天平日校时砝码采用真值还是标示值计算范围?
— 天平日校时砝码采用真值还是标示值计算范围?有没有出处?
向导@寻找流星落:
你好,采用标示值。
首先,砝码本身的存放、保管是具有特定要求的,其本身材质也较为稳定,产生变化的风险很低;
其次,从定义来说,GMP 环境下,校准 的定义可参见中国GMP指南 厂房与设备中2.6校准部分 :
校准(Calibration):是指“在规定条件下,为确定测量仪器或测量系统所指示的量值,或实物量具或参考物质所代表的量值,与对应的由标准所复现的量值之间关系的一组操作。”
另,根据JJF1001-1998《通用计量术语及定义》 ,对于真值来说,其本身是具有不确定性的。一般也会采用约定真值,即对于给定目的具有适当不确定度的、赋予特定量的值。这个约定真值,从实践看通常体现为标识值。
Q5. 生物药数字修约原则 先比较还是先修约
— 生物药数字修约原则
1:是先判断是否合格再修约至与可接受标准一致?还是先修约至与可接受标准一致再判断是否合格?
2:若需要先判断是否合格再修约,是仅对药品及中间产品 检测适用,还是对其它检测如物料 检测/公用系统检测等均适用此原则?
向导@空空如也:
《数值修约规则与极限数值的表示和判定》GB/T 8170-2008标准 中提到了两个比较方法:
4.3.2. 全数值比较法---将测试所得的测定值或计算值不经修约处理(或虽经修约处理,但应标明它是经舍、进或未进未舍而得),用该数值与规定的极限数值比较,只要超出极限数值规定的范围(不论超出程度大小),都判定为不符合要求。例如:药品或中间产品某项CQA 纯度检测结果为96.96%,若标准为≥97.0%,可以直接判定为不合格。
4.3.3. 修约值比较法---将测定值或其计算值进行修约,修约数位应与规定的极限数值数位一致。同样应用上面的例子,药品或中间产品某项CQA纯度检测结果为96.96%,先修约后为97.0%,若标准为≥97.0%,可以直接判定为合格。
由上可知,不同的比较方法,会导致不同的判定结果。
GB/T 8170-2008中规定:“4.3.1.2当标准或有关文件中,若对极限数值(包括带有极限偏差 值的数值)无特殊规定时,均应使用全数值比较法。如规定采用修约值比较法,应在标准中加以说明。”
GB/T 8170-2008中提到“4.3.4两种判定方法比较,……对于同样的极限数值,若它本身符合要求,则全数值比较法比修约值比较法相对较严格”。
《中国药典》2020版凡例中 “试验结果在运算过程中,可比规定的有效数字多保留一位数,而后根据有效数字的修约规定进舍至规定有效位。计算所得的最后数值或测定读数 值均可按修约规则进舍至规定的有效位,取此数值与标准中规定的限度数值比较,以判断是否符合规定的限度”。
E.P. 10.0版:In determining compliance with a numerical limit, the calculated result of a test or assay is first rounded to the number of significant figures stated, unless otherwise prescribed.
USP:The observed or calculated values shall be rounded off to the number of decimal places that is in agreement with the limit expression. Numbers should not be rounded until the final calculations for the reportable value have been completed.
药典中提示采用修约法比较法。由于极限数值发生一般比较少见,因此一般先修约再比较和先比较再修约的判定结果一样。
除非特殊情况下,发生极限报告数值,修约值比较法和全数值比较法两种方式可能判定结果不一样(如上述关于“纯度”的例子)。这种情况下修约值比较法虽判定合格,但建议基于风险与历史检测数据进行趋势分析,采取必要的调查(如OOT )、评估或其它适宜处理措施。
Q6. pre-IND时是否需要递交风险控制计划(RCP)?
— 请教各位老师,pre-IND 时是否需要递交风险控制计划(RCP)呢?之前看到有些公司会交,但我没有找到有相关法规说必须要交。希望有经验的老师可以多多指教,谢谢!
向导@Rich
2022年10月28日回答更新
国家药品监督管理局药品审评中心于2022年10月27日发布了《药物临床试验方案审评工作规范(征求意见稿)》 ,其中要求Pre-IND会议中提交风险管理 计划。虽然该文件还在征求意见,但是建议提交风险管理计划。
2022年10月10日原回答:
法规没有要求要交。交不交取决于你沟通交流会问的问题是什么。如果RCP是问题的支持性资料,当然就得交,如果不是,就不必交。
有种说法是Pre-IND是IND的预审评,所以最好一股脑把全套CTD 资料都交上去,让老师预审评。根据个人的项目经验,这种问法收到的答复差异性会比较大,大多数收到的回复会比较笼统,但是也会有老师给出比较具体的建议。之前记得有个药审中心沟通交流座谈会会议纪要,里面有CDE的审评老师明确表示更加喜欢明确的问题:
当然,从企业的角度来说,不管怎么样,都值得一试,胳膊肘得往内拐 。
最后,我还想提自己的两点感受和理解:
第一,沟通交流会反馈的质量如何,除了问问题的技巧之外,更重要的,还有药品的临床意义。
第二,“法规要求是最低要求”的意思是,做工作不能只盯着法规,必须思考在没有法规的情况下,我能怎么解决这个问题,想好之后再回过头拿法规来查漏补缺。世间没有人是天天按照刑法民法上的要求生活的。
【参考资料】
《药物研发与技术审评沟通交流管理办法》
《药审中心沟通交流企业座谈会-会议纪要》
识林社区是一个面向制药业从业者的互动交流平台,主要功能包括问答、文章、课程。
在这里,成百上千的识林用户提出问题,而回答问题的有其他用户,还有识林向导,识林内部团队和专家。
药品生命周期 ,是大量法规、知识、实践的结合,大家每天都会遇到大大小小的问题,但并不总能得到好的答案。
一个好的答案,不能是一句道听途说,也不仅是从“经验”出发的一句简单判断。更何况,越重要、越复杂的问题,往往并无完全正确、拿来即用的答案。
所以,优质解答,应兼具“依据”、“思考”和“建议”。读完之后,不仅可基于“建议”有所行动,也能审视答者的“思考”逻辑,还可以确认“依据”,并发散学习,真正把答案转化为自己的知识,应对未来其他问题,“鱼”与“渔”兼得。
欢迎用户登录识林社区 ,参与问答与讨论。
另外,识林小程序上线啦,随时、随地、随心进行学习与分享!搜索识林小程序,进入识林小程序吧!
识林® 版权所有,未经许可不得转载
岗位必读建议:
药品注册专员 :应熟悉本办法,负责与药审中心沟通,准备会议资料和申请。项目管理人员 :需了解会议组织流程,确保沟通交流的合规性。研发团队 :应明确沟通交流的目的和议题,准备相应的技术资料。文件适用范围:
要点总结:
沟通交流形式 :明确了面对面、视频、电话会议或书面回复等沟通形式,并鼓励使用电话会议。会议类型 :区分了Ⅰ类、Ⅱ类和Ⅲ类会议,针对不同研发阶段和问题进行沟通。申请与审核流程 :规定了沟通交流会议的申请条件、资料提交要求和项目管理人员的审核职责。会议准备与召开 :强调了会议资料的准备、会议的组织和召开流程,以及会议纪要的撰写和存档。延期与取消条件 :说明了会议延期和取消的情形,以及相关的通知和处理流程。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA:确保药品生产和检验过程符合《中国药典》四部的要求。 注册:在药品注册过程中,需参照《中国药典》四部的通用技术要求和品种正文。 研发:在药品研发阶段,需关注药典对原料药和药用辅料的技术规定。 生产:在生产过程中,需遵循药典对生产工艺和贮藏条件的规定。 文件适用范围:
文件要点总结:
药典结构与效力 :《中国药典》由四部分组成,四部主要包含通用技术要求和药用辅料标准,其规定具有强制性,替代旧版药典标准。药品标准构成 :药品标准由品种正文、凡例和通用技术要求共同构成,确保药品质量符合规定。药用辅料要求 :药用辅料需符合相关法律、法规及药典通则,品种正文详述了药用辅料的技术规定。检验方法与限度 :所有品种应按规定方法检验,方法适用性需确认,限度数值包括上限和下限。标准品与对照品 :用于药品质量控制的标准物质,需经过技术审定,确保其满足既定用途。以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。 QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA:负责确保实验室操作符合质量控制要求,监督取样、留样、检验等流程。 研发:在设计质量标准和分析方法时,需遵循本文规定。 生产:在取样和留样过程中,应遵守本文的详细规定以保证产品质量。 文件适用范围:
文件要点总结:
实验室职责与布局: 明确了质量控制实验室的职责、布局原则和要求,以及人员的组织架构和资质要求。取样与留样管理: 规定了取样过程的控制和留样的定义、量、储存要求及记录。物料和产品检验: 强调了检验要求,包括待检样品核对、检验、记录和报告书的编制。委托检验管理: 阐述了委托检验的原则、应用范围、职责和工作流程。质量标准建立: 详细说明了质量标准的设计与制定、审核与批准流程。以上仅为部分要点,请阅读原文,深入理解监管要求。