首页
>
资讯
>
周年盘点 – 识林学习者过去一年最关注的法规和资料
出自识林
周年盘点 – 识林学习者过去一年最关注的法规和资料
2019-05-10
识林APP上线一年来,已为上万名制药人提供专业法规和技术资料的检索和学习的知识服务。百万次的学习量,是应对快速变化的国内外法规环境和技术要求的坚实基础,也是追求知识型产业,学习型企业和求知型人才的识林用户的步伐见证。
那么一年来,制药人都在学什么呢?本文回顾了一年来制药同仁最关注的内容。
杂质和生物分析方法是工艺开发和产品质量热点
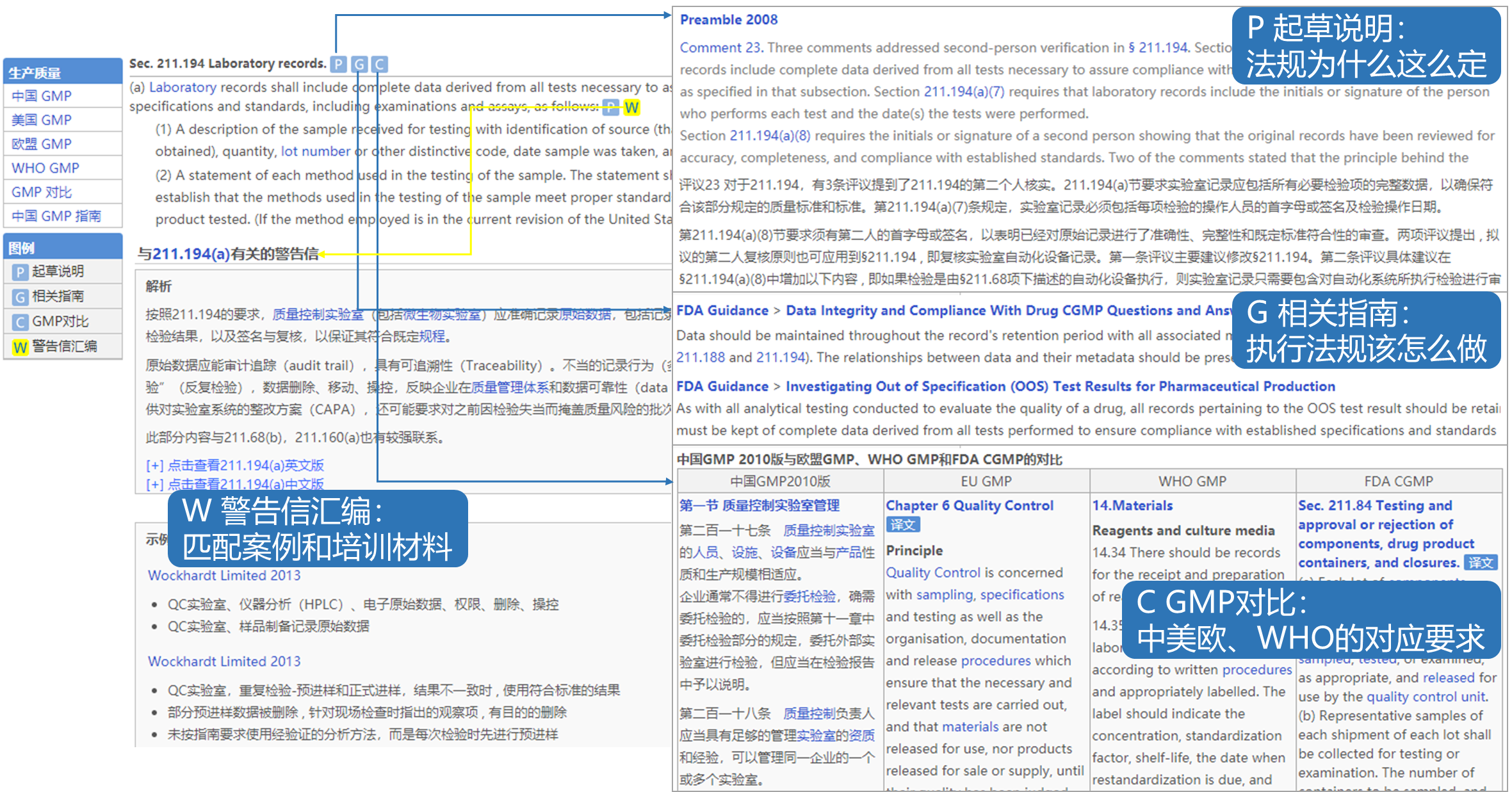
没有太多意外,带有法规解读、对应指南、检查缺陷和中英文翻译比对的中美GMP使用和查询非常方便,一直是识林中最实用的注释性学习资料。尤其是FDA的官方法规解读(Preamble),识林发布了完整中译版,是学习法规实质和精髓的无价资料。
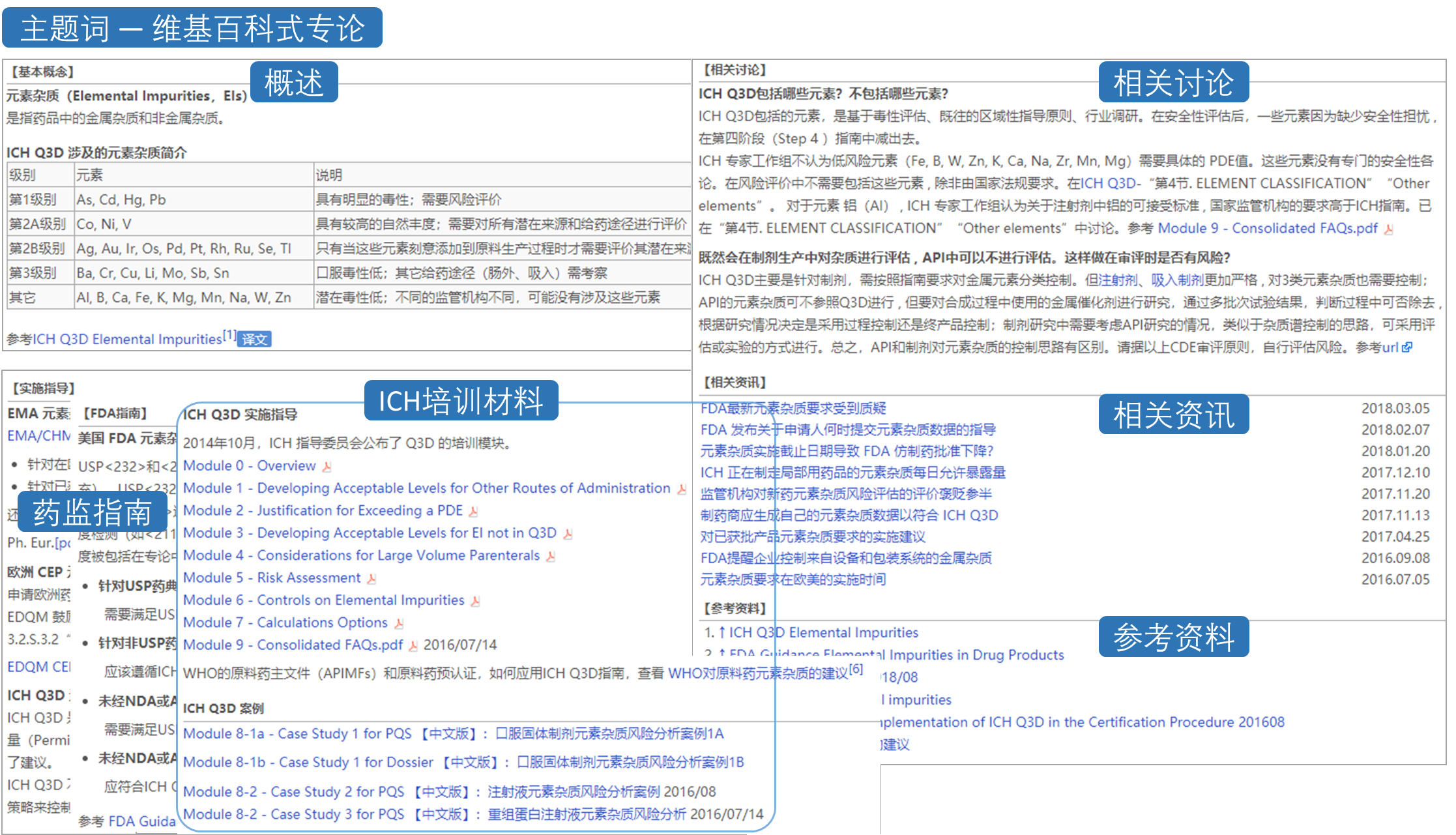
ICH系列指南是第二类最受关注的法规,受法规和缬沙坦事件的影响,杂质和基因毒性 相关的指南学习量最大。
除了全部ICH指南的翻译,识林还有超过800个专业主题的链接,从概念原理,各国法规要求,实施指导,实操案例和官方培训材料等方面系统深入学习。
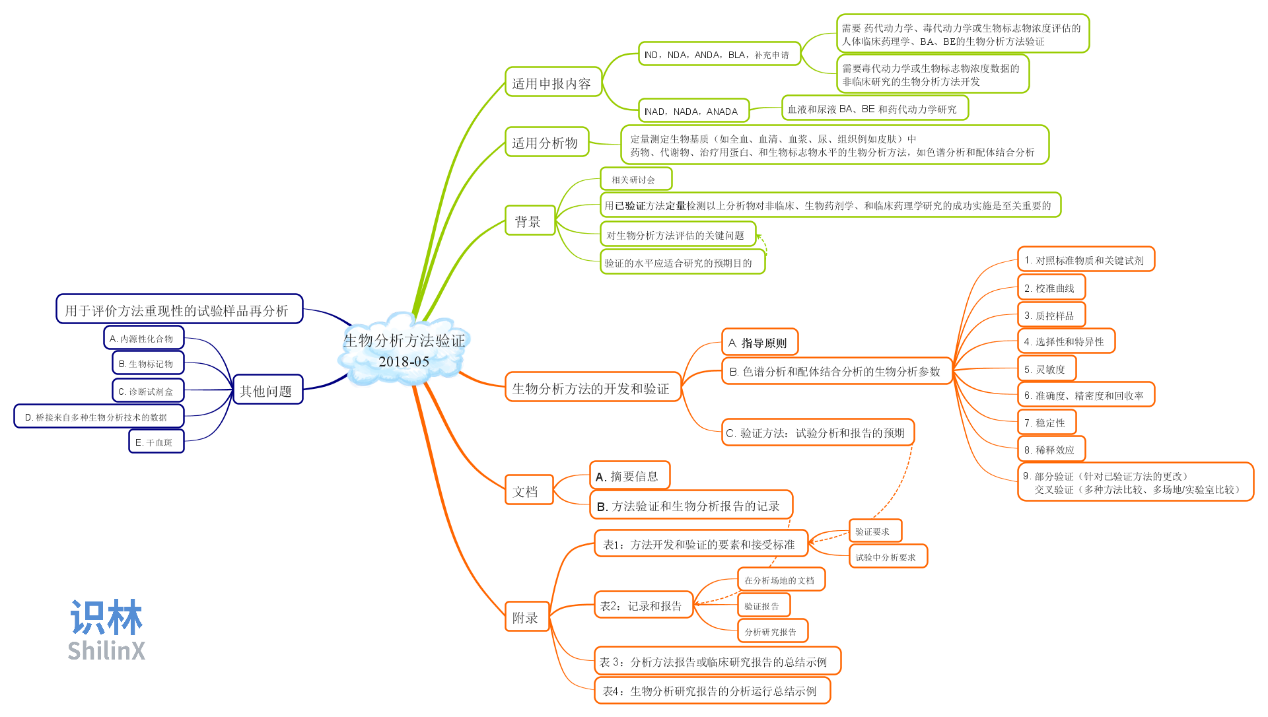
此外,FDA 2018年5月的新版生物分析方法验证 指南,药品管理法修正案,欧盟的GMP和上市前问答,也是学习量最大的法规指南,识林也添加了指南导读提升学习效率。下图是生物分析方法验证的导图。
错过了什么: 合规程序手册(CPGM) ,对FDA批准前检查 、BE 临床检查、API检查、GMP 检查、无菌检查 及生物制品检查的思路,检查重点和问题做了详细阐述,还有MAPP和SOPP包含了化学药和生物药 的申报和评价的流程和要点,但学习量没有应有的多。【无菌企业自检清单 — FDA检查员手册的168个问题】
识林案例和工具提供工作任务解决方案
过去一年,识林团队完成了近百个对比解读、案例和任务工具包,学习量大和实用性强有很大关系,尤其是任务工具包,从实际工作任务流程出发,细化到每个环节的技术要点,并且关联到所需的法规政策文件和专业主题词。最受关注的内容包括:
错过了什么: eCTD , 吸入式制剂 ,生物等效性 ,冻干 ,国际危机管理 ,统计学应用 、监管科学 等数十个深度研究专题,还有生物制剂工艺、工艺 变更 等更多专题在开发中。
检查缺陷和警告信一封封单独学习效率不高
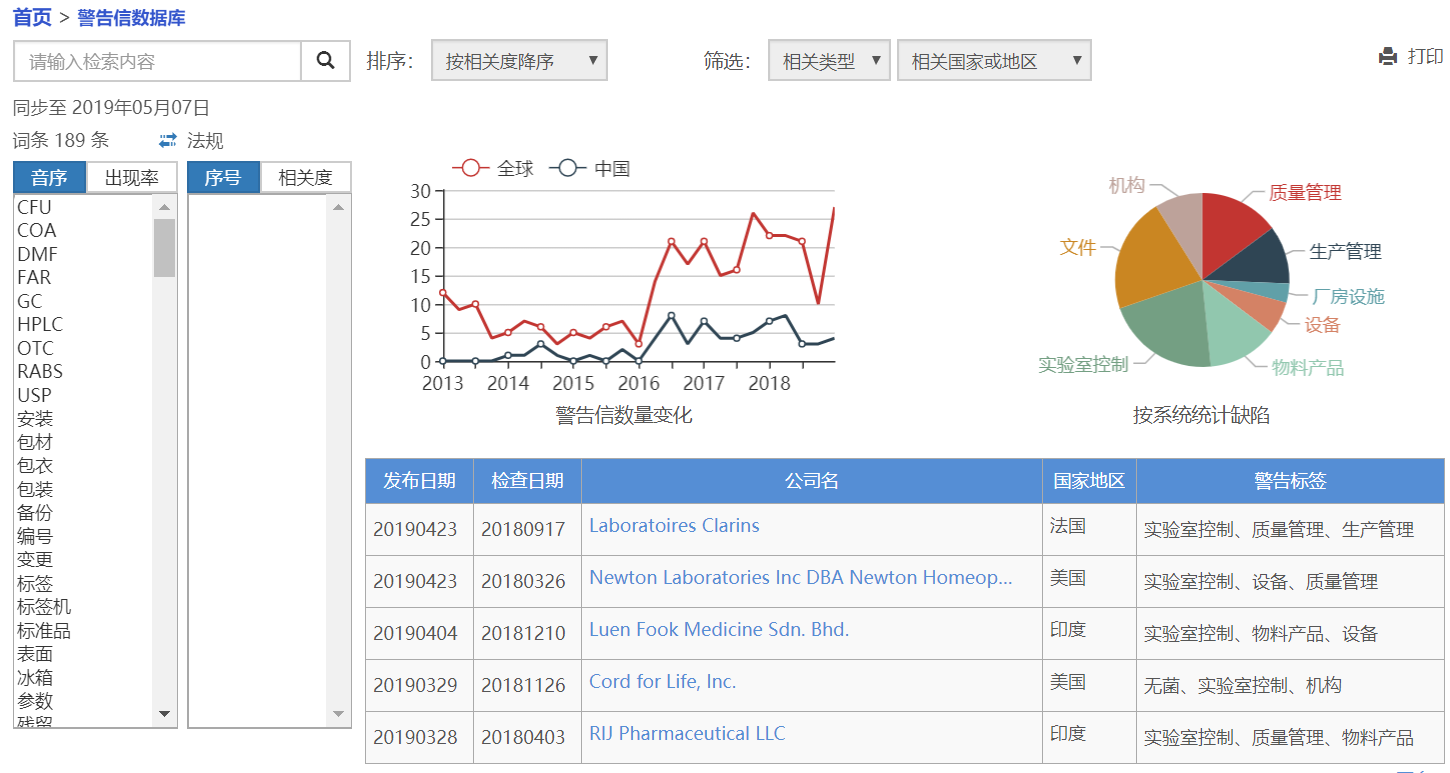
美国FDA进口禁令 清单,CNDA 药品飞行检查 江西国药有限责任公司 20180509,NMPA 药品飞行检查 珠海亿邦制药股份有限公司20180624,FDA WL Zhejiang Huahai Pharmaceutical 20181129 中文翻译和FDA WL Zhuhai United Laboratories Co. Ltd. 20180627,是第三类学习量大的资料。尽管识林翻译了全部的FDA药品GMP警告信,并收录了大量483缺陷,检查员信息,欧盟检查缺陷和国内飞检和各省检查缺陷数据,逐个学习数以千计的资料并不是识林推荐的方法。
错过了什么: 警告信数据库 提供了按缺陷条款、关键词、公司、国家、产品类型等检索方式和各种组合和关联分析,能找到相关缺陷的完整清单,相关缺陷 和关联程度,可用于风险评估、体系自检、培训迎检等,国家局和各省检查缺陷数据库也即将上线。
识林还有哪些变化?
识林已有超过25000篇全球法规和实践学习资料,并能够24小时同步更新的国内(国家局、相关部委、主要省局等)和全球(FDA、EU、WHO、PIC/S、ICH等)药监法规和技术资料动态。过去一年识林新建和更新了近万篇内容,覆盖研发注册在内的全产品生命周期 。专业能力诊断测评版块已经在APP上线,包含28个课程模块,500多个视频的在线教学模块 也即将在识林APP上线。
适用岗位及工作建议:
QA(质量保证) :必读。应根据指南更新内部质量控制流程,确保生物分析方法验证的合规性。研发(R&D) :必读。在新药研发过程中,需遵循指南要求,对生物分析方法进行严格的验证和优化。临床(Clinical) :必读。在临床试验中,应确保生物样本分析的准确性和可靠性,符合指南规定。注册(Regulatory Affairs) :必读。在准备IND、NDA、ANDA、BLA等申请时,需参考指南确保提交的数据符合FDA要求。文件适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
QA(质量保证):负责监督和确保药品生产过程符合M7(R1)指南要求,确保基因毒性杂质控制在可接受水平。 R&D(研发):在新药研发过程中,需评估和控制合成过程中产生的基因毒性杂质,确保药物安全性。 临床研究:在临床试验阶段,需根据M7(R1)指南评估药物中的基因毒性杂质,确保受试者安全。 注册:负责药品注册文件的准备和提交,需根据M7(R1)指南提供基因毒性杂质的评估和控制策略。 工作建议:
QA:定期审查生产流程,确保基因毒性杂质的控制措施得到有效实施,并符合M7(R1)指南要求。 R&D:在药物合成和制剂开发阶段,主动识别和评估潜在的基因毒性杂质,采取控制措施以降低风险。 临床研究:在设计临床试验方案时,考虑基因毒性杂质的风险评估,并在试验报告中包含相关数据。 注册:在药品注册文件中明确说明基因毒性杂质的评估结果和控制策略,确保符合监管要求。 适用范围:
要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
关键日期:本文件为 ICH Q1A(R2) 指南的当前步骤 4 版本,日期为 2003 年 2 月 6 日。该指南已被 ICH 专家工作组制定,并已根据 ICH 流程由监管方咨询。本指南在 ICH 流程的第 4 步被推荐采纳。
适用范围:本文件适用于新药物物质和产品的稳定性测试,包括化学药品、生物制品、疫苗和中药等。适用于注册分类(创新药或仿制药、生物类似药、原料药等)和监管市场(如中国、美国、欧盟等)。适用于 Biotech、大型药企、跨国药企、CRO 和 CDMO 等企业类型。
适用岗位:本文件对药品研发、注册、质量保证(QA)、生产、市场等岗位的工作带来变化,特别是对稳定性研究和注册申报的岗位是必读。
文件要点总结:
稳定性测试目的:为证明药物物质或药物产品在各种环境因素(如温度、湿度、光照)影响下随时间变化的质量,并建立药物物质的复验期或药物产品的货架期及推荐储存条件。 稳定性测试条件:基于对 EC、日本和美国三个地区气候条件影响的分析,世界被划分为四个气候区域 I-IV,本指南针对气候区域 I 和 II。 药物物质稳定性测试:包括压力测试、批次选择、容器封闭系统、规格、测试频率、储存条件、稳定性承诺、评估和标签声明等。 药物产品稳定性测试:包括光稳定性测试、批次选择、容器封闭系统、规格、测试频率、储存条件、稳定性承诺、评估和标签声明等。 稳定性数据包:本指南定义了新药物物质或药物产品注册申请所需的核心稳定性数据包,同时允许足够的灵活性以适应由于特定科学考虑和被评估材料特性可能出现的不同实际情况。 以上仅为部分要点,请阅读原文,深入理解监管要求。
法规指南解读:ICH_Q3A(R2)_Impurities_in_New_Drug_Substances
适用岗位(必读):
研发(R&D):负责新药物质的化学合成和杂质分析。 质量管理(QA):确保新药物质的质量符合ICH指南要求。 注册(Regulatory Affairs):准备注册文件,确保包含所有必要的杂质信息。 工作建议:
研发:在新药物质的合成过程中,识别和控制有机、无机杂质及溶剂残留。 质量管理:建立和验证分析程序,确保能够检测和量化杂质含量。 注册:在注册文件中详细报告杂质含量,包括批次分析和稳定性研究数据。 文件适用范围:
文件要点总结:
杂质分类: 明确了有机杂质、无机杂质和溶剂残留的分类,并指出了不包括在本指南中的杂质类型。报告和控制理由: 强调了在新药物质合成、纯化和储存过程中实际和潜在杂质的总结,以及实验室研究的重要性。分析程序: 要求注册申请中包含验证过的分析程序,以检测和量化杂质。批次杂质含量报告: 要求提供用于临床、安全和稳定性测试的新药物质批次的分析结果。规格中杂质列表: 新药物质规格应包括杂质列表,基于商业生产批次中发现的杂质选择。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位必读建议:
QA(质量保证部门):应全面理解并执行GMP原则,确保所有相关活动符合规定要求。 生产部门:必须遵循文件中关于生产操作、设备维护和清洁、校准以及计算机化系统的规定。 研发部门:在API的初期研发阶段,应考虑GMP原则以确保产品质量。 临床部门:在临床试验阶段使用API时,应遵守特定的GMP实践,确保临床试验材料的质量。 文件适用范围:
文件要点总结:
质量管理体系 :强调所有参与制造的人员应负责质量,建立并实施有效的质量管理体系。人员 :确保足够的合格人员参与API的制造,并进行定期培训。建筑与设施 :建筑设计应便于清洁、维护,并最小化污染风险。生产设备 :设备的设计、建造和维护应确保不影响API的质量,并定期校准。文件和记录 :所有相关文档应按照书面程序准备、审核、批准和分发,确保记录的完整性和可追溯性。材料管理 :建立材料接收、鉴定、隔离、存储、取样、测试和批准或拒绝的书面程序。生产和过程控制 :监控生产步骤的进展并控制过程变异性,确保API的一致性。验证 :包括验证政策、文件、资格认证、过程验证方法和周期性审查已验证系统的程序。变更控制 :建立正式的变更控制系统,评估可能影响中间体或API生产和控制的所有变更。投诉和召回 :记录并调查所有质量相关的投诉,并制定召回程序。以上仅为部分要点,请阅读原文,深入理解监管要求。