|
首页
>
资讯
>
国内药政每周导读:我国疫苗监管通过WHO评估,药典委2022年拟提高的标准,仿制药目录57批发布
出自识林
国内药政每周导读:我国疫苗监管通过WHO评估,药典委2022年拟提高的标准,仿制药目录57批发布
2022-08-29
【CMC与仿制药】
8.25,【药典委】关于做好2022年度国家药品标准提高(第二批)工作的通知
药典委公布了2022年度国家标准提高课题的清单,企业CMC开发以及生成质量部门有必要关注,了解国家标准下一步的变化。
其中,品种标准提升涉及到:
- 复方天麻颗粒
- 健脾颗粒
- 参苏感冒片
- 达沙替尼和片
- 枸橼酸托法替布和片
- 乳酶生
- 肾上腺素注射液
- 盐酸西那卡塞和片
- 依非韦伦和片
通用技术要求涉及到:
- 含马兜铃酸中成药检测方法及指导原则研究
- 剧毒、管制试剂试药删除与替换课题
- 《红外光谱集》编制
- 宿主细胞残余DNA片段大小分布检测方法的研究
- 0861残留溶剂测定法的修订
- 药品无菌保障指导原则的研究(二阶段)
- 磷脂类辅料相变温度通用测定方法的建立及作 为辅料功能性指标的可 行性研究(第二阶段)
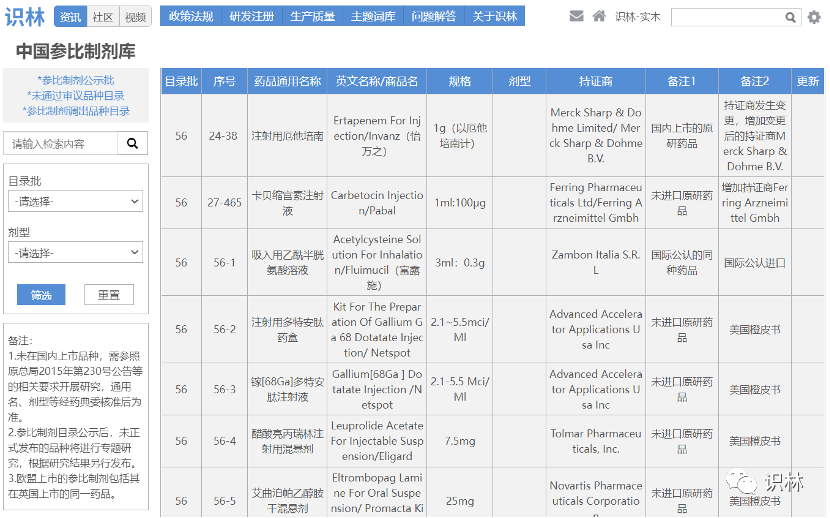
8.26,【NMPA】关于发布仿制药参比制剂目录(第五十七批)的通告(2022年第38号)
参比制剂已公示六十一批,正式发布五十七批。
可至识林“中国参比制剂库”查阅。
【注册,审评,审批】
8.26,【黑龙江省】公开征求《关于药品上市后场地变更实施办法》(征求意见稿)意见
黑龙江局发文的整体导向,仍然是在职权范围内,为省内企业提供各种便利。
对于境内场地变更,按照2021年1月《药品上市后变更管理办法》(试行)的规定,除了涉及到工艺、处方、质量标准等其他注册管理事项变更的,均由省局审批。
要点如下:
- MAH引入外省品种办B证,免于提交转出方省局同意受托意见。这一条多个省局都已提及。
- 原生产地址已废止的,可以先变更,但承诺不得实际生产(许可证上还要备注清楚),还需再做研究。
- 质量对比研究的做法,可以“选择质量标准、仿制药参比制剂、市售主流或通过一致性评价的品种为对照开展质量对比研究”。
- 实在无法做质量对比研究的中药独家品种,经风险评估,有机会免除。
- 同剂型整体变更,选一个品种的一个规格3批验证,即可办变更。当然,品种上市前,该做的研究和验证还是不能少。
- MAH仍然委托原厂生产的,可用1年内的GMP符合性检查免去变更的现场检查。
可见,黑龙江文的特点,是把“场地变更”和“生产上市”分开来处理,使得企业可在新场地开展必要的变更研究和申报。
识林汇总了此类“省局药品上市后变更管理实施细则”,可点击链接查阅。
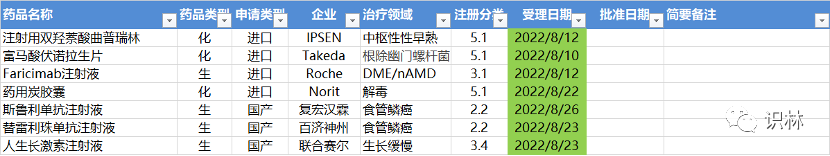
8.28,CDE 药品审评报告和说明书更新,其中包括下列国产新药
注:仅列出国产创新药和改良型新药,供参考。企业用户可至识林“CDE 药品审评报告”按发布时间查阅最新的上市药品信息。
| 受理号
| 药品名称
| 药品类型
| 注册分类
| 企业名称
| 承办日期
|
| CXSS2000047
| 贝伐珠单抗注射液
| 治疗用生物制品
| 3.3
| 上海复宏汉霖
| 2020-09-09
|
| CXSS2000046
| 贝伐珠单抗注射液
| 治疗用生物制品
| 3.3
| 东曜
| 2020-09-05
|
| CXSS2000027
| 贝伐珠单抗注射液
| 治疗用生物制品
| 2
| 海正;贝达
| 2020-06-17
|
【政策与监管综合】
8.23,【NMPA】我国疫苗监管体系通过世界卫生组织评估
2022年8月23日,世界卫生组织(WHO)宣布中国通过疫苗国家监管体系(NRA)评估。
WHO将通过疫苗监管体系评估作为采购该国疫苗产品的前提,即只有国家监管体系通过评估,该国企业才能申请WHO疫苗产品预认证,并列入联合国等国际组织采购清单。此外 , 通过评估也是其他国家注册和采购他国疫苗产品的重要参考。
我国疫苗监管体系已于2011年、2014年先后两次通过评估,在2022年7月迎来了WHO升级评估标准后的新一轮全面评估。
【新药批准和报产】
8.22-8.28,NMPA未发布新药批准,CDE受理7个NDA
注:仅列出新药(包括改良型新药和生物类似药)的上市申请和批准上市信息。在“以临床价值为导向”的背景下,申请上市以及获批的品种,其适应症、临床研究策略、注册路径,都值得业界关注和分析。
识林-实木
识林®版权所有,未经许可不得转载。
适用岗位及工作建议: - QA(质量保证):必读。确保公司变更管理流程符合法规要求,监督变更实施过程。
- RA(注册事务):必读。负责变更申请的提交、审批和备案工作,确保变更文件的合规性。
- 生产管理:必读。负责变更实施的现场管理,确保生产过程符合变更要求。
适用范围:
本文适用于中国境内的化学药、生物制品、疫苗、中药等药品的上市后变更管理,包括创新药、仿制药、生物类似药、原料药等注册分类,适用于Biotech、大型药企、跨国药企、CRO和CDMO等各类企业。 要点总结: - 变更管理责任:强调持有人为变更管理的责任主体,需建立变更控制体系。
- 变更类别:明确审批类、备案类和报告类变更的管理要求,确保变更不影响药品安全性、有效性和质量可控性。
- 持有人变更管理:规定变更持有人后,需保持药品一致性,变更后持有人负责药品全生命周期管理。
- 生产场地变更管理:要求变更生产场地后,药品质量需与原药品一致,重大变更需报批。
- 变更程序和监督管理:规定审批、备案、报告的具体程序,强调监管部门的监督检查职责。
以上仅为部分要点,请阅读原文,深入理解监管要求。 适用岗位: - QA(质量保证):必读,需确保残留溶剂测定符合中国药典要求。
- QC(质量控制):必读,负责执行残留溶剂的测定方法。
- R&D(研发):必读,需了解残留溶剂的测定方法以优化生产工艺。
- 注册:必读,需在药品注册文件中包含残留溶剂的合规性信息。
工作建议: - QA:监控残留溶剂测定流程,确保符合GMP要求。
- QC:执行测定方法,记录数据,确保数据完整性和准确性。
- R&D:在新药开发中考虑残留溶剂的控制策略,以满足药典标准。
- 注册:在药品注册资料中明确残留溶剂的控制和测定方法。
适用范围:
本文适用于化学药品,包括创新药和仿制药,由中国药典发布,适用于大型药企、Biotech公司、跨国药企以及CRO和CDMO等企业。 文件要点: - 残留溶剂定义与分类:明确了残留溶剂的定义,并根据毒性分为三类,规定了各自的残留限度。
- 测定方法:强调使用气相色谱法进行残留溶剂测定,包括色谱柱的选择和系统适用性试验。
- 供试品与对照品溶液制备:详细规定了供试品和对照品溶液的制备方法,包括顶空进样和溶液直接进样。
- 测定法与计算:介绍了三种测定方法(毛细管柱顶空进样等温法、程序升温法和溶液直接进样法)及其计算方式。
- 特殊注意事项:包括含氮碱性化合物的测定、检测器选择、顶空平衡温度和时间的选择等。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |