首页
>
资讯
>
国际药政每周概要:ICH Q8、Q9和 Q10问答;MHRA 选出五种创新 AI 技术试点;WHO 逐步取消生物制品质量控制动物试验指南;FDA 加速审批指南更新,最佳指南规范报告和计划,等
出自识林
国际药政每周概要:ICH Q8、Q9和 Q10问答;MHRA 选出五种创新 AI 技术试点;WHO 逐步取消生物制品质量控制动物试验指南;FDA 加速审批指南更新,最佳指南规范报告和计划,等
2024-12-12
【监管综合】
12.06 【ICH】Q9(R1)附件1:关于Q8、Q9和Q10的问与答(R5)
本文件主要涉及以下方面的问答:一般性说明;质量源于设计 (设计空间 、实时放行 检测、控制策略 );药品质量体系 ;ICH 质量指南对 GMP 检查实施的影响;知识管理 ;软件解决方案。
2024 年 10 月 30 日 ICH 对该文件批准了以下修订:
- 鉴于第 12、13、15、18 和 19 页上 ICH Q8 、Q9 和 Q10 的实施,删除现在被认为过时的文本并重新措辞问答;
- 在第 4、10、12 和 17 页上进行少量添加,以解决文档中的小内容空白;
- 进行少量编辑以提高文档的可读性,例如第 6、10、12、15 和 18 页上所示的编辑。
12.04 【MHRA】MHRA 选出五种创新人工智能(AI)技术参与旨在改变监管方式的试点计划
MHRA已为AI Airlock试点计划选择了五项创新技术,旨在更好地了解如何监管人工智能 (AI)驱动的医疗器械 。AI Airlock是一种监管“沙盒”研究,生产商可以在其中探索如何最好地收集证据,这些证据以后可用于支持其产品的批准。这是在MHRA监督下在虚拟或模拟环境中完成的。
被选中用于 AI Airlock 并不构成监管机构的批准。该试点的结果将于2025年公布,将为未来的AI Airlock项目提供信息,并影响未来的英国AI医疗器械指南。
选定的五种技术包括:
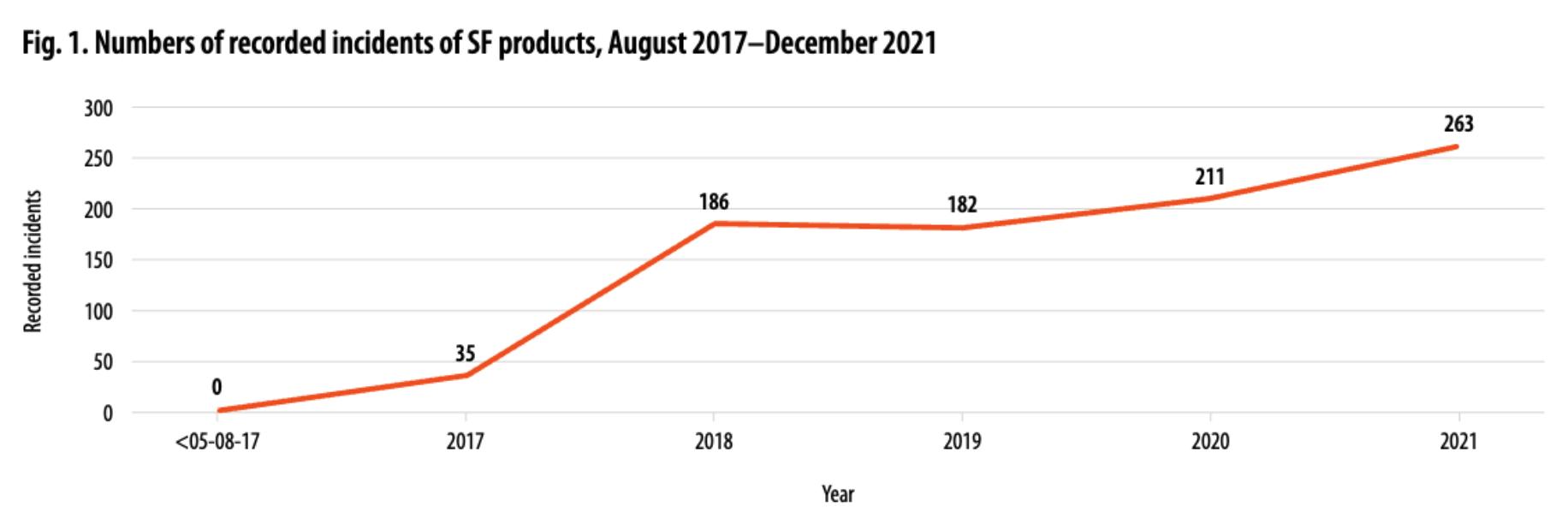
12.03【WHO】全球劣质和伪造医疗产品监测系统:2017年8月至2021年12月的行动报告
这份关于 2017-2021 年期间劣质和伪造(SF)医疗产品全球监测和监控系统 (GSMS) 的报告提供了洞察,描述了挑战并提出了预防、检测和应对的策略和行动。
报告表明,SF 医疗产品报告事件数量惊人地增加,包括各种基本药物 ,例如抗菌药物、肿瘤药物和疫苗。在所涵盖的期间内,记录了 877 起事件,年均增长率为 36.3%。
鉴于未来 SF 医疗产品的数量将持续增加,该报告呼吁采取一致行动。
建议包括加强监管系统、加强国家法律框架、建立可持续报告系统、改善监管部门和执法部门之间的协调、提高公众意识和促进国际合作。
12.02 【WHO】关于逐步取消生物制品质量控制动物试验的指南
动物试验在生物药品的开发中具有长期而重要的作用,可提供有关其作用机制,安全性和有效性的关键信息。 在许多情况下,这些方法在批准后继续用于监测产品质量和/或安全性,作为生产商和国家控制实验室执行的质量控制 过程的一部分。 近来,生物制品 质量控制的非动物试验技术在开发和实施方面取得了重大进展,部分原因是应用了3R原则,并认识到这些方法优于现有的常规质量控制动物试验。
该指南为开发人员、生产商和监管机构提供关于逐步淘汰用于生物制品质量控制的动物试验的指导。指南包括将非动物方法与现有动物方法进行比较以建立相关性具有挑战性或不适当的示例;但是,它不包括对含量 测定验证本身的详细讨论。
12.02 【FDA】FDA 最佳指南规范报告和计划
12.02 【FDA】FDA 与利益相关方沟通的最佳实践:报告征求意见稿
这份报告作为 2023 年国会拨款的一部分发布,报告称,FDA“大幅增加了每年发布的指南文件数量”,但补充说明“发布指南文件使 FDA 的建议变得透明且易于理解,促进了一致性,并避免了不必要的费用和产品开发延误。”
生物制药行业和共和党议员此前曾将 FDA 使用指南文件视为过度监管行为,尤其是与每年发布的少得多的正式规章数量相比,因为即使指南不具有法律约束力,行业通常也必须遵循指南。
FDA 并未反对这一说法,但其在报告中表示,指南可能对较新或规模较小的公司尤其重要,“这些公司与 FDA 打交道的经验可能较少,聘请外部法律顾问或专家的资源也较少。”
报告还明确指出,FDA 认为应该“更新其已有 20 多年历史的良好指南实践法规。”
参见资讯:FDA 发布报告详述有关指南使用的最佳实践
12.06 【EMA】药品质量问答:第一部分新增术语“打开胶囊服用的颗粒”和“打开胶囊服用的口服粉末”问答
12.06【WHO】WHO 宣布修订晚期 HIV 疾病管理指南
12.06【FDA】FDA 综述:2024年12月06日
12.05【WHO】乙肝和丙肝检测服务规划中的优先事项:新的操作指南
12.05【WHO】以人为中心的乙肝和丙肝检测服务规划中的优先事项 - 操作指南
12.05【EMA】植物药委员会质量起草小组三年工作计划
12.05【FDA】血液机构的重要信息:关于网络安全弹性
12.04 【FDA】COVID-19 药品和非疫苗生物制品紧急使用授权 内容更新
12.04 【FDA】Catherine Gray 博士谈潜在的虚假或误导性处方药推广的识别和报告
12.04【EU】欧盟委员会推出罕见病跨境医学讨论新平台
12.03【FDA】FDA 综述:2024年12月03日
12.03【WHO】国际卫生条例(IHR)脊髓灰质炎应急委员会第四十次会议的声明
12.02 【FDA】关于第19届国际药品监管机构会议的思考
12.02 【EU】欧盟委员会新指定三个欧盟公共卫生参考实验室
12.02 【WHO】WHO 药物信息 - 第38卷第3期
【注册、审评、审批】
12.06 【FDA】指南定稿 用于计划 CDER 申报的生物研究监测(BIMO)检查的 NDA 和 BLA 内容电子递交的标准格式
指南阐述了 FDA 药品审评和研究中心(CDER)希望从主要研究中获取哪些类型的数据。FDA 表示,“为了按照 CDER 良好审评管理原则和《处方药使用者付费法》(PDUFA )所涵盖产品的实践实现审评绩效目标,CDER 通常在申请审评流程的早期(即,在立卷决定和审评规划阶段)启动检查规划。
CDER 的检查规划包括选择临床研究者场地和其它受监管实体进行现场检查,以及准备 CDER 提供给检查和调查办公室 (OII)检查员的任务备忘录和背景资料,这些检查员负责执行 FDA 的 BIMO 检查。”CDER 使用指南中描述的数据和信息来规划 BIMO 检查,包括 (1) 促进及时确定检查场地,(2) 确保 OII 检查员能够获得进行 BIMO 检查所需的信息。
参见资讯:FDA 定稿规划生物研究监测(BIMO)检查的电子申报指南
12.05【FDA】指南草案 关于严重病症的加快审评计划 - 药品和生物制品的加速批准
指南反映了最新立法对加速审批 计划的更改。根据《2023 年综合拨款法案》(Consolidated Appropriations Act of 2023,CAA),国会要求对 FDA 的加速审批计划进行修改,赋予 FDA 新权力来设定确证性试验 的条件,并对于未能证明临床获益的产品建立新的撤销程序。
FDA 指出,“本指南草案涉及加速审批流程,包括授予加速审批(例如,讨论终点、证据标准、确证性试验和其它加速审批条件)和撤回加速审批。与 2014 年定稿指南 相比,变化包括对新终点的早期咨询、及时进行确证性试验、确证性试验的其它方面以及加速审批的加快撤销。其它变化包括修订标题和编辑性更改以更清晰易懂,以及更新 FDA 的参考资料和联系信息。”
国会还要求 FDA 修改其撤销加速批准的程序,以确保未能满足批准后预期的药物可以迅速撤出市场。FDA 指出,如果申办人未能满足其批准后研究要求、确证性研究未能显示临床获益、其它证据表明该药物未能满足安全性和有效性要求,或者申办人使用虚假或误导性信息来推广该药物,FDA 可能会使用快速程序撤销加速批准。
参见资讯:FDA 发布加速审批修订指南,反映加快撤销程序
12.05【FDA】MAPP 5019.2 药品和生物制品注射剂的包装净含量的评定
该手册适用于单剂量和多剂量西林瓶装的药品,包括已批准用于固定剂量给药方案的制剂和已批准按体重或体表面积给药的制剂。FDA 表示,正确的容器净含量 对于患者的正确给药、最大程度地减少污染和限制药品浪费非常重要。
滥用灌装到西林瓶和其它包装类型中的注射用药品,包括不安全的处理和注射技术,导致药品污染,并增加患者之间血源性疾病传播的风险。可能导致患者和医疗保健提供者不安全的处理和注射行为的一个因素是不适当的净容器容量大小,导致:
需要使用多个西林瓶给一个患者用药,当混合来自不同西林瓶的物料时,由于不适当的无菌技术,可能导致污染 风险增加。
FDA 在 MAPP 中指出,产品质量评估办公室(OPQA I)或OPQA II或OPQA III质量审评员将在产品开发过程中尽早通知研究性新药申请(IND )的申办人,拟定的药品净容器容量应为适用于预期用途和剂量。
参见资讯:FDA 更新注射剂容器净含量程序政策手册
12.05【EMA】寻求关于传统植物药的科学支持和建议的申请人指南
12.05【FDA】MAPP 5200.7 Rev.1 由立卷审评处审核 ANDA 增补和补充申请
12.04【FDA】FDA 加速批准 zenocutuzumab-zbco 用于非小细胞肺癌和胰腺腺癌
12.04【FDA】FDA 批准度伐利尤单抗用于局限期小细胞肺癌
12.04 【EMA】2024-2025向 EMA 递交孤儿药申请的期限和相应有效申请的 COMP 时间表
12.04【EMA】上市许可持有人和申请人指南:如何支付 EMA 收取的费用
12.04 【PMDA】药品 已获批产品清单 新增 FY 2024、All (April 2004 to September 2024) 更新
12.04【PMDA】药品 审评报告 新增 Vyloy
12.04 【WHO】世卫组织宣布首次通过一款结核病诊断检测工具预认证
12.03【EU】2025年工作计划 - 成员国卫生技术评估(HTA)协调小组(HTACG)
【研发与临床】
12.04 【EMA】关于需要修订用于体重控制的药品的临床评价指南关于儿童体重控制的附录的概念性文件
由于新体重管理药品的批准以及当前临床实践指南的变化,EMA 提议更改儿童体重管理附录。EMA 提议更新附录介绍,以反映当前的科学知识和发展。
其它计划的修订涉及儿童超重/肥胖的定义和临床研究 设计。在讨论该定义时,EMA 表示,目前的附录引用了体重指数(body mass index,BMI)相关指标来确定儿童的超重和肥胖。EMA 认为当前方法存在缺点。“BMI 应该根据年龄和性别进行调整,并且指标可能并不理想,因为不能以个性化的方式直接反映肥胖或过度肥胖的后果。其它工具可能有助于更好地描述药物治疗的目标人群。”
在试验设计方面,EMA 列出了计划改变的五个方面。EMA 正在考虑取消三个月的导入期要求,称这可能会阻止人们参与试验。其还在考虑“根据预期的目标人群”改变入组和排除标准。EMA 还在研究现有的对于停止治疗后六个月观察期的要求。EMA 表示,“停止治疗可能有其缺点,可能不会增加很多信息。因此,将考虑停药后是否需要随访以及随访时间的长短。”
参见资讯:欧盟就儿童体重控制临床试验监管征求反馈意见
12.06【EMA】NASH/MASH 临床试验中在肝活检中使用基于人工智能的非酒精性脂肪性肝炎组织学测量以确定疾病活动性的确认意见草案
12.05【EMA】皮下和/或肌肉注射用普通人免疫球蛋白(SCIg/IMIg)的临床研究指南
12.05【EMA】皮下和肌肉注射用普通人免疫球蛋白(SCIg/IMIg)的核心产品特性总结(SmPC)指南
12.05【FDA】药物试验快照:VYLOY
12.04【WHO】用于预防呼吸道合胞病毒疾病的单克隆抗体和相关产品的非临床和临床评价
12.04 【EMA】A 型和 B 型血友病非替代疗法的临床要求指南
12.04【FDA】药物试验快照:AQNEURSA
12.04【FDA】药物试验快照:MIPLYFFA
12.03【FDA】药物试验快照:NIKTIMVO
【安全性】
12.04【EMA】关于在扩展 EudraVigilance 药品词典(XEVMPD)中以电子方式提交人用研究用药品信息的指南
12.03【WHO】使用队列事件监测进行猴痘疫苗的安全性监测:WHO 方案
【GxP 与检查】
12.05【FDA】483 印度 Fresenius Kabi Oncology Limited
12.03【FDA】警告信 美国 Applied Therapeutics, Inc.
12.03【FDA】警告信 美国 Han C. Phan, M.D.
12.03【FDA】警告信 中国 浙江优全护理用品科技股份有限公司
12.03 【FDA】警告信 韩国 LCC Ltd.
12.02 【EudraGMDP NCR】捷克 Elkoplast Slusovice s.r.o.
【仿制药和生物类似药】
12.06【FDA】仿制药计划月度和季度活动报告(2025财年)
12.03【FDA】即将发布的仿制药研发特定产品指南 内容更新
【医疗器械】
12.03【FDA】指南定稿 人工智能驱动的器械软件功能的预定变更控制计划的上市申报建议
12.02 【EU】MDCG 2024-15 关于在没有欧洲医疗器械数据库(EUDAMED)的情况下发布临床研究报告及其摘要的指南(2024年11月)
【其它】
12.03【EDQM】2024年11月欧洲药典委员会第180次会议结果
12.02 【BPOG】计算机建模的关键术语和缩写词汇表
识林® 版权所有,未经许可不得转载。