|
首页
>
资讯
>
欧盟无菌附录(草案)变化详解
出自识林
2018-08-27
欧盟资深专家Ian Thrussell在IPEM课堂与大家分享无菌法规和检查经验,识林将整理详情文章,敬请期待。
欧盟于2017年12月发布了《GMP附录1:无菌药品的生产》新草案,这是一份全新的无菌附录,并不是旧版的修订版。正如起草工作组的负责人Andrew Hopkins所说,草案和现在版本,无论从语言、架构等其他方面都是全新的版本。而且格式也是完全不一样。反映出在这个时期监管方面的需求,以及其他国家和地区在监管方面的贡献。例如:
- 现在共有269个条款(与2007年版的127个相比),不考虑许多子条款,也没有扩大引言和范围
- 100个与现有条款没有直接联系的新条款
- 上一版本中的14个条款在更新中未以任何形式出现
- 来自上一版本的至少70个条款已经更新,可能至少对某些制药商产生一些影响
- 原先的127个条款中,只有约40个条款没有实质性改变
变与不变:整体导读
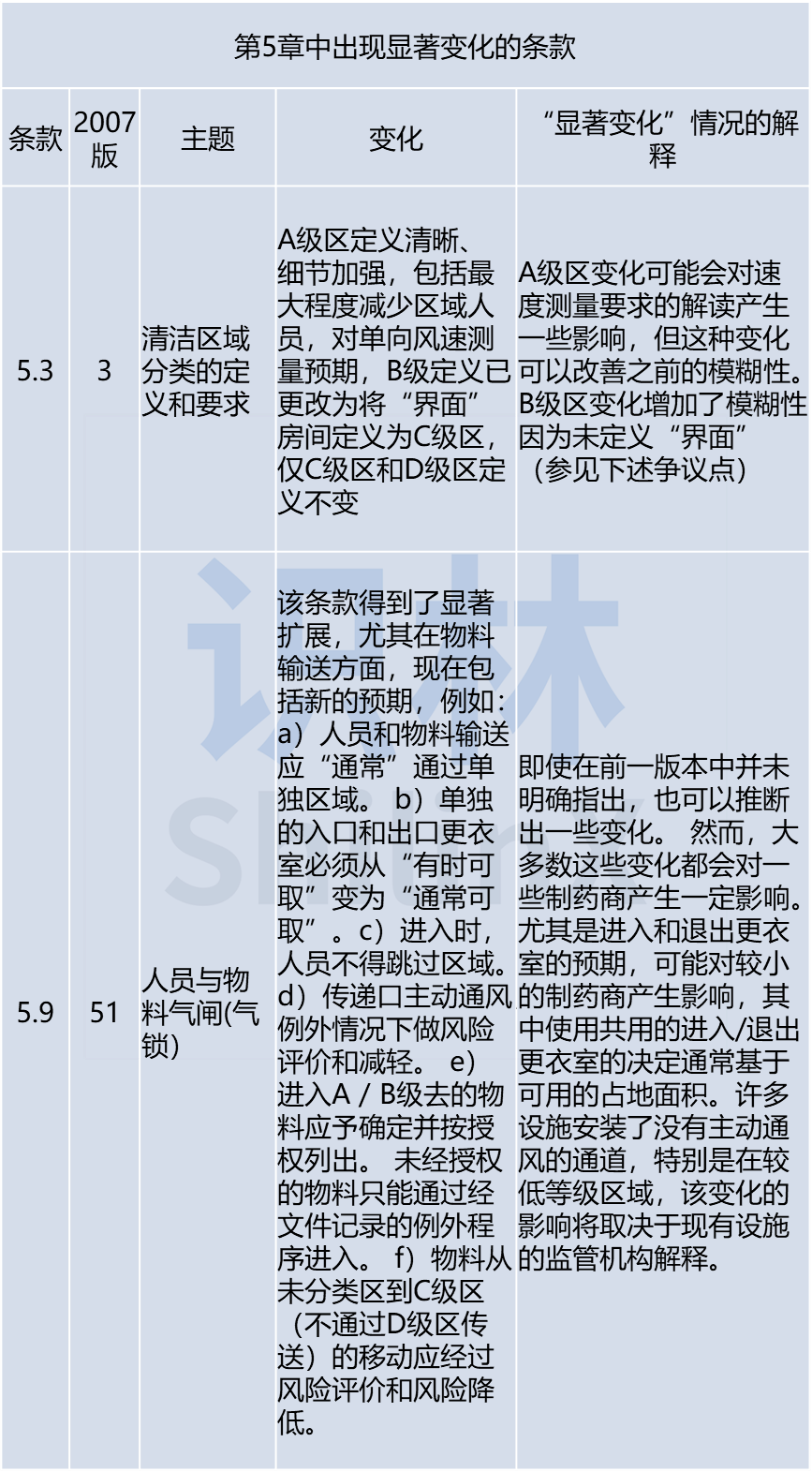
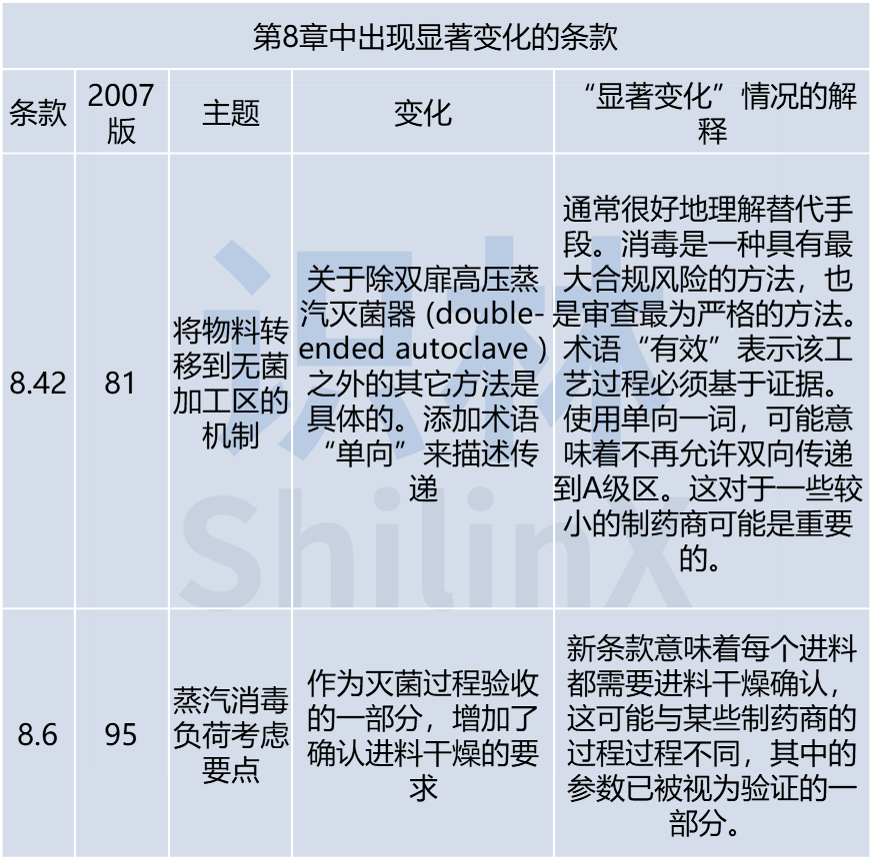
总的来看,在新版本中至少有70条,会对现在的药厂有影响,只有40条几乎保持不变,也就是说有三分之二内容被修订了,或者有大幅的改动。目前的重点集中在质量风险管理方法,对质量风险管理(QRM) 预期的更新是这一变化的关键原因,新文件中完全融入了风险管理概念。提及了一些新主题,包括:一次性使用技术、无菌操作员资质确认、对不同主题的风险管理应用、洁净间表面清洁认证、工艺用水系统、关键设施、密闭制造系统。以及提供了预计将在污染控制策略中涵盖的要素的详细清单,污染控制应讲求“战略”,希望公司对污染源有目的的进行研究,以及了解控制机制。
本附录的概念报告(concept paper)中指出,就资源或成本而言,预期不会对行业产生不利影响。因为大多数变革在很多制药场地已经存在,并且已经成为现行GMP的一部分。最有可能引发讨论和评论的关键变革领域包括:
- 对正式、整体性的污染控制策略的明确预期。
- 对反映整个场地策略的正式文件或卷宗的隐形预期。
- 用于无菌制造的最低程度的污染控制。
- 关键区域的洁净间分类(超出ISO要求)可能需要额外的要求。
- 在所有分类区(包括D级区)强制要求HEPA/ULPA过滤的潜在意图。
- 显著增强了对目视检查的预期。
逐条点评:欧盟资深专家Ian Thrussell本色解读
尽管新版本有望消除以前版本的模糊性和不准确性,但仍然存在一些问题:
- 将生物负载限制与过滤器效率联系起来的要求,如果从字面上解释,通常会导致限制,将成为一个完全失控的解决方案制造过程。
- 尽管多年来一直是一个主要的混淆话题,但关于气闸(气锁)最终级的声明保持不变。
- 对A级条件和A级进风之间的差异仍然存在一些混淆,增加了另一个术语A级环境导致更加复杂。
术语表中A级进风的定义并未完全缓解模糊性。
作者:识林-柏
识林®版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
必读岗位及工作建议: - QA(质量保证):应密切关注GMP附件1的更新,确保公司生产流程与新指南保持一致。
- 生产:需了解新指南对无菌药品生产的具体要求,以优化生产流程。
- 注册:在药品注册过程中,应考虑新指南对文件和流程的影响。
- 研发:在新药开发阶段,应将新指南的要求纳入产品设计和工艺开发中。
文件适用范围:
本文适用于欧盟/欧洲经济区成员国及药品检查合作计划(PIC/S)参与机构的无菌药品生产。涵盖的药品类型为无菌药品,注册分类包括创新药和仿制药,发布机构为EMA和PIC/S,企业类别包括Biotech、大型药企、跨国药企等。 文件要点总结: - 更新需求:鉴于技术和GMP实践的变化,特别是ICH Q9和Q10指南的采纳,需要更新GMP附件1。
- 技术与风险管理:新指南将明确ICH指南中风险管理和质量体系概念在无菌药品生产设施、设备和工艺设计中的应用。
- 国际一致性:修订将考虑与国际要求的一致性,确保与欧盟或PIC/S其他药品指南文件的连贯性。
- 新技术推广:鼓励使用新技术以提高产品质量,同时确保对现有产品的不利影响最小化。
- 清晰度提升:纠正历史不准确之处,消除模糊性,提供更清晰的GMP期望解释。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |