|
首页
>
资讯
>
阿达木单抗的 BE 试验经验总结
出自识林
2020-09-04 识林
9月3日,信达生物的阿达木单抗注射液获国家药监局批准,成为继百奥泰和海正药业之后第三个国产阿达木单抗生物类似药。
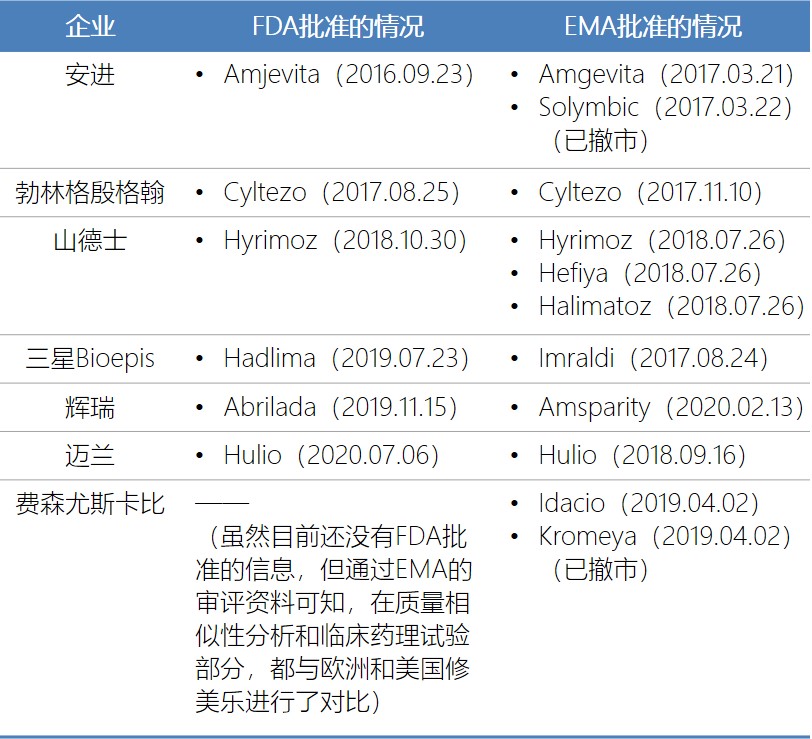
艾伯维的修美乐(阿达木单抗)于2002年12月31日首次获得美国FDA批准上市,2010年2月26日首次国内获批进口。修美乐自2014年开始,已经连续6年成为全球药品销售冠军,2019年的销售额接近200亿美元。
FDA已批准了6个公司的阿达木单抗生物类似药(几乎所有企业采用的都是欧美双报的开发申报策略),EMA除了FDA批准的这6家公司的产品,还有一个来费森尤斯卡比的阿达木单抗生物类似药,这个产品虽然还没有获得FDA批准,但通过EMA公布的审评资料可知,该公司在进行生物类似性研究时,同时比较欧洲和美国的修美乐产品。
临床药理学研究在生物类似产品研发中发挥着关键作用。这些研究是证明拟议生物类似产品和参照品之间生物类似性的循序渐进过程的一部分。这些研究可能有助于证明产品之间没有临床意义上的差异。这些研究可能可以解决在药学研究评价之后仍然残留的不确定性,可能会增进支持生物类似性证明的证据整体性,也可能有助于采用具选择性和针对性的方法设计任何建议的后续临床研究来支持生物类似性证明。
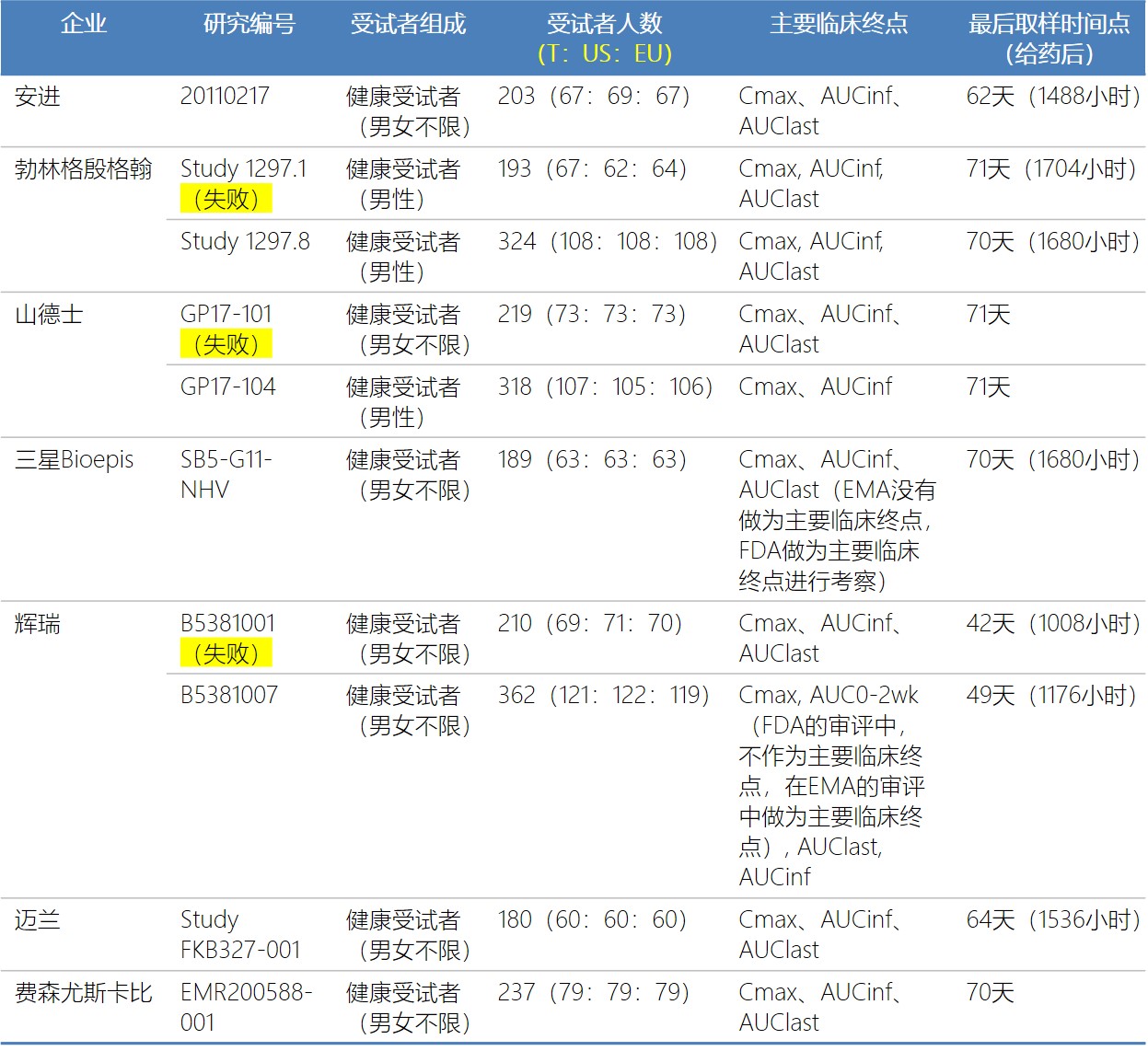
回顾这些阿达木单抗生物类似药的临床药理试验,基本的设计相同,均采用健康受试者,单剂量皮下注射给药(40 mg),双盲随机3臂试验。但具体的细节设计上,各企业又存在一定差异:
受试者:
虽然各企业都选择了健康受试者进行试验,但更细致的要求有所差别,主要体现在性别、年龄、体重、BMI等要求上。
企业估算的样本量大小差异比较明显,单臂受试者的数量从60人到120人。
随机化:有些试验在随机化前进行了体重分层。
主要临床终点:FDA在2016年发布的生物类似药临床药理学评价指南指出,对于单剂量研究,在计算AUC时应比较AUCinf,对于皮下注射研究,应选择Cmax和AUC做为共同研究终点。所有企业的主要临床终点中均包含Cmax和AUCinf,大多数企业还将AUClast也纳入了主要临床终点的评价范围。
约定的等效范围:90%置信区间【0.80,1.25】。
最后取样时间点:大部分企业选择的最终取样时间点在60-70天左右(只有辉瑞除外,2次试验的最终取样时间点分别未42和49天)。
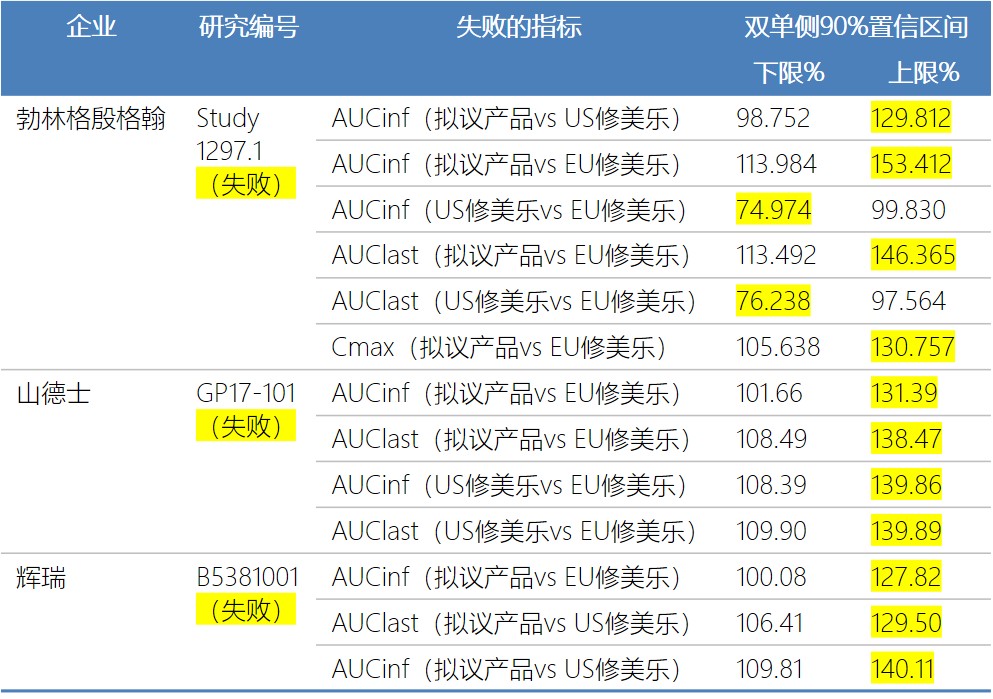
7家公司有3家公司的第一次试验以失败告终,没有达到约定的相似性临床终点。
3个失败的临床药理试验的主要临床终点AUCinf和AUClast超出了约定的等效范围导致了试验失败,勃林格殷格翰的Cmax也超出了等效性的上限。
这3家公司分析了第一次临床药临试验的结果,调整了试验方案进行了第2次临床药理试验,均成功。
勃林格殷格翰
在第一次Study 1297.1失败后,勃林格殷格翰总结失败原因为,高的总体变异度,体重对暴露剂量的影响,参比制剂和试验药品蛋白浓度的差异,未预料到的欧洲修美乐暴露量低。
在进行第二次Study 1297.8时,公司:
增加了受试者人数(从193人增加到了324人)
严格规定了注射部位仅限于小腹
在方案中预先定义将体重作为协变量纳入ANCOVA分析
山德仕
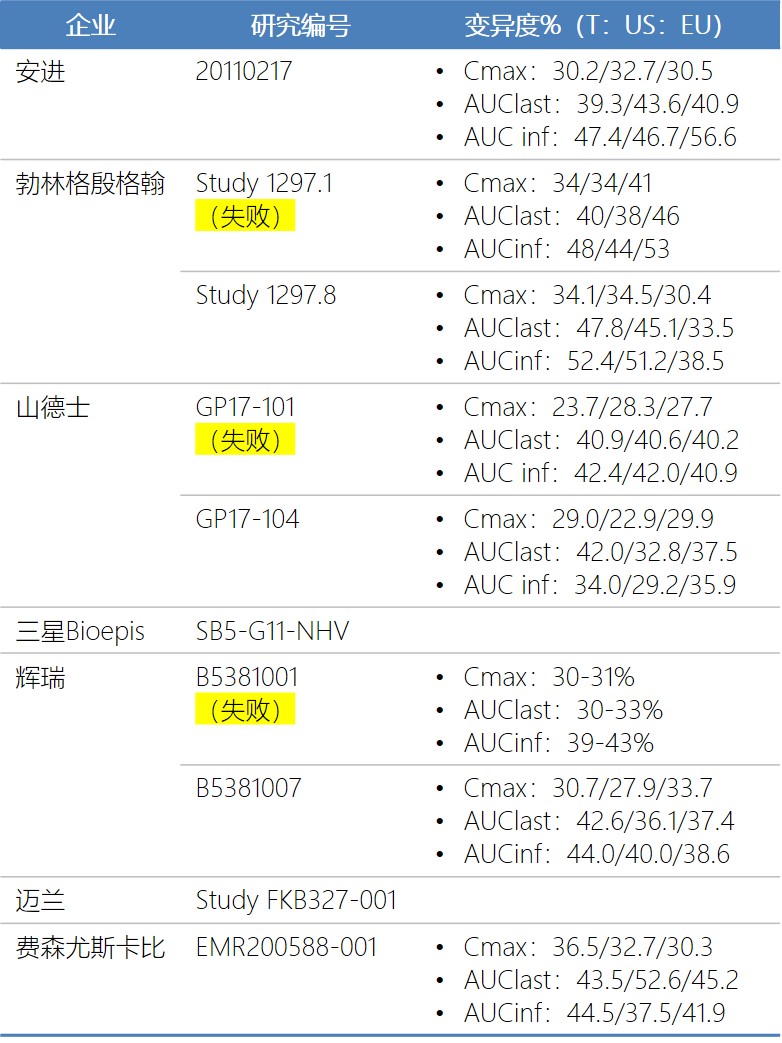
山德仕分析了第一次GP17-101失败的原因时没有发现于产品或试验相关的单个根本原因,分析观察到试验的AUClast实际变异性大于40%,高于申请人预先估计的31%的变异度。山德仕在对GP17-101进行数据回顾分析后,认为ADA的形成对阿达木单抗的暴露也有影响。
在进行第二次GP17-104试验时,山德仕:
将受试者人数从219人调整到318人
还将严格了受试者人群,从健康成年受试者调整为健康男性受试者
在随机化前对体重进行了分组
还将主要临床终点从3个(Cmax、AUCinf、AUClast)减少为2个(Cmax、AUCinf)
在试验方案中约定对ADA阳性和ADA阴性受试者的AUClast和AUCinf描述性比较
辉瑞
辉瑞分析第一次临床药理试验(Study B5381001)失败的原因,归结于受试者间的变异度(最大45%)高于估算样本量时假设的变异度(30%)。
在进行第二次临床药理试验(Study B5381007)时,对试验设计进行了以下调整:
受试者人数从210人(单臂70人)调整到360人(单臂120人)
血液样品收集的时间范围从42天调整到49天
在随机化分组时,增加了根据体重分层
严格了受试者入组标准:年龄范围从18-55岁缩小到18-45岁;BMI指数从17.5-30.5 kg/m2缩小到19.0-30.5 kg/m2;最低体重要求从50 kg调整到60 kg
临床药理试验的失败主要原因归结于变异度,3家企业在估算样本量时,都低估了变异度。回顾已经完成的10个临床药理试验,Cmax的变异度在22.9-36.5%之间(多数在30%左右),AUClast的在30-52.6%之间(多数在40%左右),AUCinf的在29.2-56.6%之间(多数在40%以上)。
因为推测低估了变异度导致估算的样本量小,所以3个企业都根据第一次失败试验计算出的变异度,重新估算了样本量大小,增加了受试者人数。
为了降低了变异度对试验的影响,企业还通过严格了受试者人群:
山德仕将受试者人群限定为健康男性
辉瑞则缩小了受试者的年龄范围
企业认为体重可能是增加变异度的一个因素,因此BI增加了协变量体重分析,山德仕和辉瑞在进行随机化前增加了根据体重分组,辉瑞还缩小了受试者的体重范围。
取样时间范围也可能影响AUCinf的评价。AUCinf代表的是开始到无限时间的血药浓度曲线下面积,计算方式为AUCinf= AUClast+ Ct/kel(Ct<在最后可测量时间点的浓度>除以kel<消除速率常数>),是基于适当的方法计算的。一般要求AUClast的覆盖至少达到AUCinf的80%以上,取样时间覆盖足够长的血药浓度时间曲线,可以提供可靠的药量程度估计。在终末对数线性阶段期间需要至少3到4个样本,以便可靠地估计终末速率常数,这是可靠估算AUCinf所需的。因此,辉瑞将取样时间范围从42天增加到49天(但仍然少于其他企业的取样时间范围)。
生物大分子的复杂性使得生物类似药的研发之路包含了很多不确定性。阿达木单抗生物类似药的临床药理试验的失败率较高,希望其他企业的宝贵经验,可以减少失败率,降低成本缩短时间,点滴积累起的惊喜让明日之路走得更平坦。
作者:识林-木兰
识林®版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
|