首页
>
资讯
>
CIRS 报告纳入 NMPA,展示国际化新药在中国的获批趋势
出自识林
CIRS 报告纳入 NMPA,展示国际化新药在中国的获批趋势
2025-09-22
9月16日,国际组织监管科学创新中心(Centre for Innovation in Regulatory Science, CIRS)发布报告《追踪六大全球主要市场已批准药品在中国的上市情况》 (CIRS RD Briefing 102–Tracking Availability in China of Medicines Approved in Six Key Global Markets),追踪分析 2019 - 2023 年间在澳大利亚、加拿大、欧洲、日本、瑞士和美国六个全球主要药品市场获批的新活性物质(New Active Substances, NAS)在中国的可及性情况。
报告数据截至2025年1月,而CIRS 2015-2024年六家监管机构新药批准趋势简报 还是在前不久的8月份才发布的,这解释了为何不包含2024年六大市场数据。CIRS从2012年开始每年发布批准分析报告,这次将NMPA纳入。尽管是延迟发布单独的报告,但可以预期明年报告将囊括NMPA在内的七家监管机构。
除了时效性,报告结论是基于CIRS的统计口径和计算规则,可能与当前最新情况不尽相符,仅供参考。
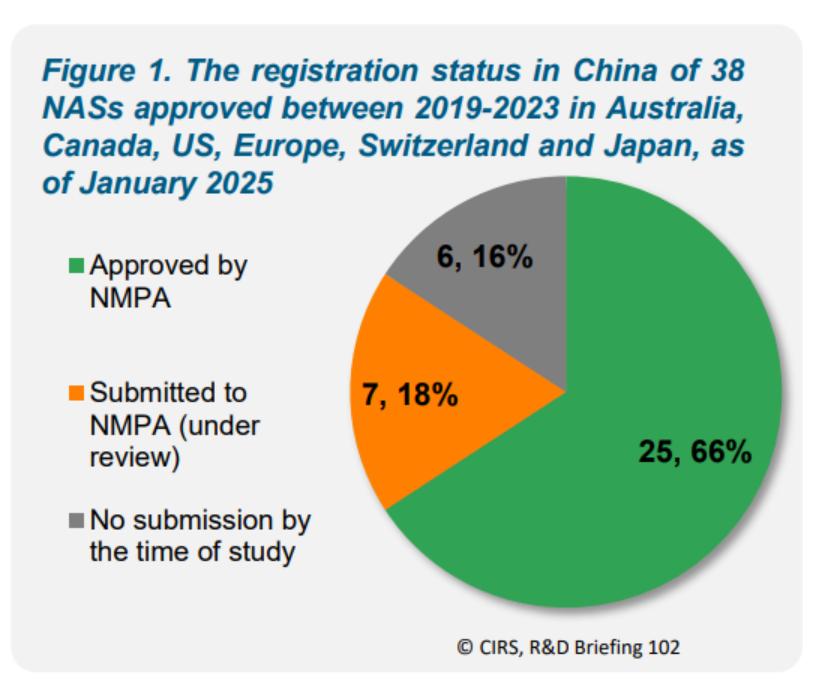
报告指出,2019 - 2023 年间,六大监管机构共批准 38 种 NAS,其中 25 种(66%)已获NMPA批准,7 种处于审评中,6种未见申报。这 25 种获七家监管机构批准的 NAS被CIRS定义为 “国际化药品”(internationalised medicines)。从药品类型来看,64% 为化学药品,36% 为生物制品 ;治疗领域方面,60% 为抗肿瘤和免疫调节剂。
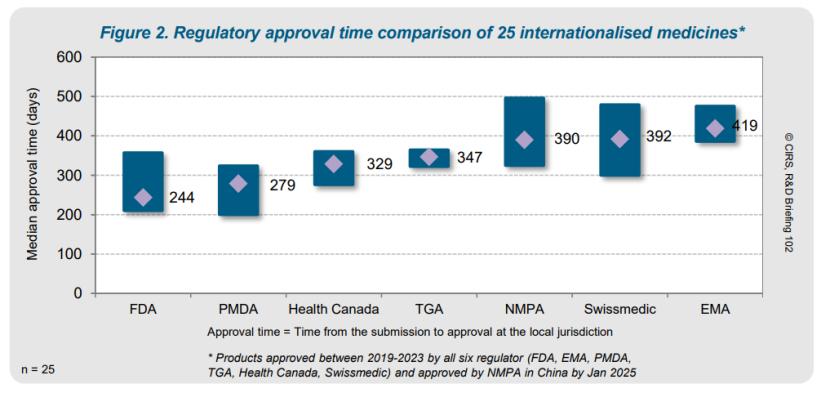
审评时间线和加速审批 方面,FDA 审批时间中位数最短,为244天。FDA、PMDA 和 NMPA 最常使用加速审评 路径,这使 NMPA 审评时间中位数(390天)与其他监管机构相当,但由于提交间隔大,导致推进上市时间(rollout time)延长。25种 NAS 中,12种获 NMPA 优先审评 ,其中两种同时被认定为突破性疗法,四种获附条件批准。对比FDA批准的NAS中,76%经优先审评获批,其次为日本PMDA的56%、EMA的48%。
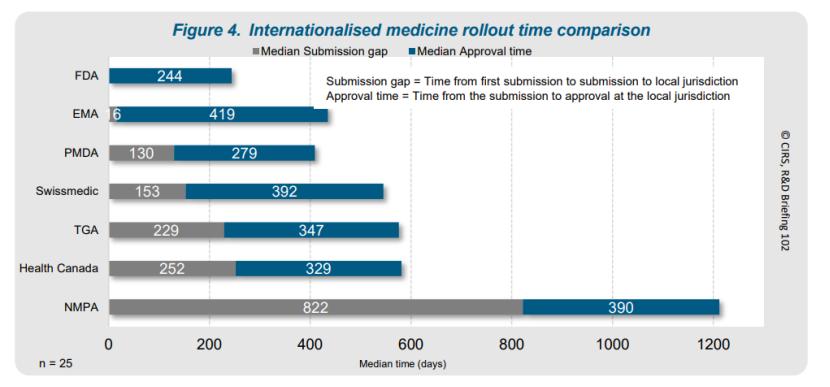
在提交模式和全球推进上市时间方面,FDA 通常是首个受理新药提交的机构,占比 84%,其次是 EMA。向 PMDA 的提交模式较为多变(按上述2024年最新报告是727天,略低于NMPA)。中国提交间隔中位数最长,达822天,常为最后提交地区。
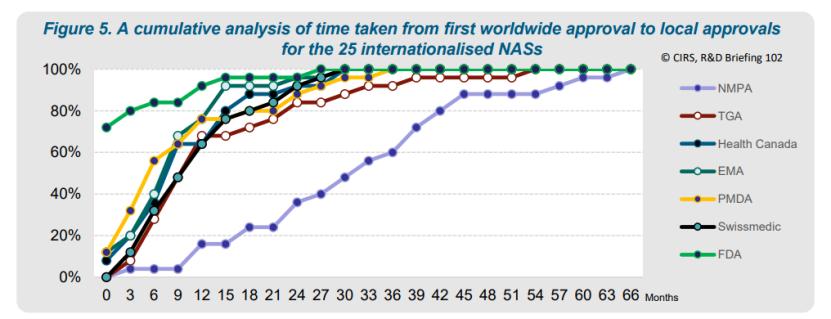
有些企业效率本就较高,11种NAS在两年至三年内向七家机构提交,6种在一年内完成全球提交。FDA在70%的国际化药品中率先批准或在首次全球批准后一个月内批准。其他市场也相对迅速,中国批准上市的延迟还是归因于企业提交策略。
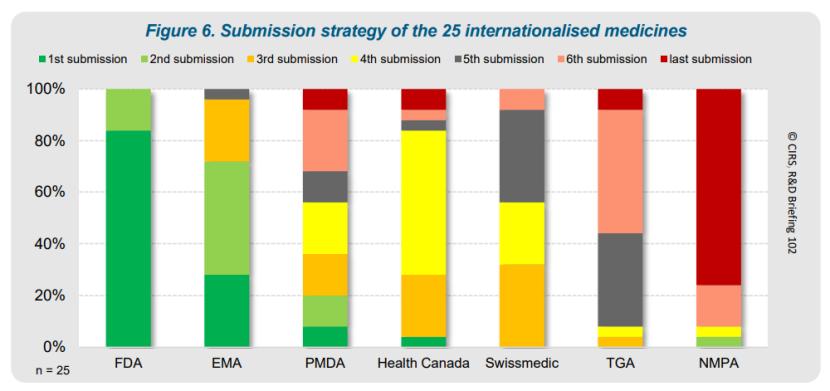
虽然这个数据不甚理想,但其实不同药品在中国的提交时间存在较大差异,表明各企业策略有所不同。如下图,大片的红色意味着企业最后才在中国提交,但也有少数绿色显示个别企业较早提交。与之相对,瑞士和TGA总是从第三家提交开始。
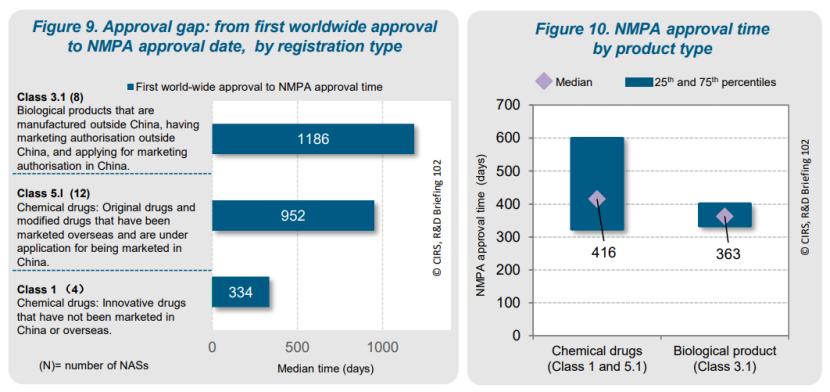
报告进一步分析了不同注册类型的NAS的审批时间差距。1类创新化学药(编者注:该报告采用了NMPA注册分类 )的审批时间最短,从首次全球批准到NMPA批准的中位时间为334天。这是因为1类药品在获得全球首次批准之前就提交NMPA了,中位提前时间为151天。相比之下,5.1类化学药和3.1类生物制品的提交时间较晚,分别在首次全球批准后527天和838天提交给NMPA。尽管提交时间较晚,但NMPA的审批时间相对稳定,化学药(1类和5.1类)和生物制品(3.1类)的审批中位时间分别为416天和363天。
报告还探讨了公司规模的影响。大型研发公司(定义为2021年研发投入超过30亿美元的公司)通常比非大型公司更快地将药品提交给NMPA。具体来说,大型公司的提交时间中位数为706天,而非大型公司为1021天。在审批时间方面,大型公司开发的NAS虽然审批时间稍长,但波动较小,中位审批时间为393天,而非大型公司为330天。这表明大型公司在全球监管策略中更倾向于早期考虑中国市场,而NMPA的审批流程一贯比较稳定。
识林-实木
识林® 版权所有,未经许可不得转载