首页
>
资讯
>
EMA 2023 年报,详述审评和监督检查成就
出自识林
2024-06-12
欧洲药品管理局(EMA)于上个月发布其 2023 年度报告 ,详述了 EMA 在去年一年的监管活动,报告采用全新的布局和新的互动功能,可供深入了解 EMA 的战略举措以及 2023 年工作重点。
报告和 24 个附件总共 200 多页内容,回顾了 EMA 2023 年人用药和兽药审评和监督重点以及 EMA 在三大战略领域的关键成就:抗癌药、数据驱动的药品监管以及透明度和沟通。今年的报告有两种格式可供查看,除了传统的 PDF 版本外,新增了允许用户过滤数据并与数据进行交互的电子版本(https://www.ema.europa.eu/en/annual-report/2023/index.html url ),阅读和检索起来更加方便。
药品和监管活动
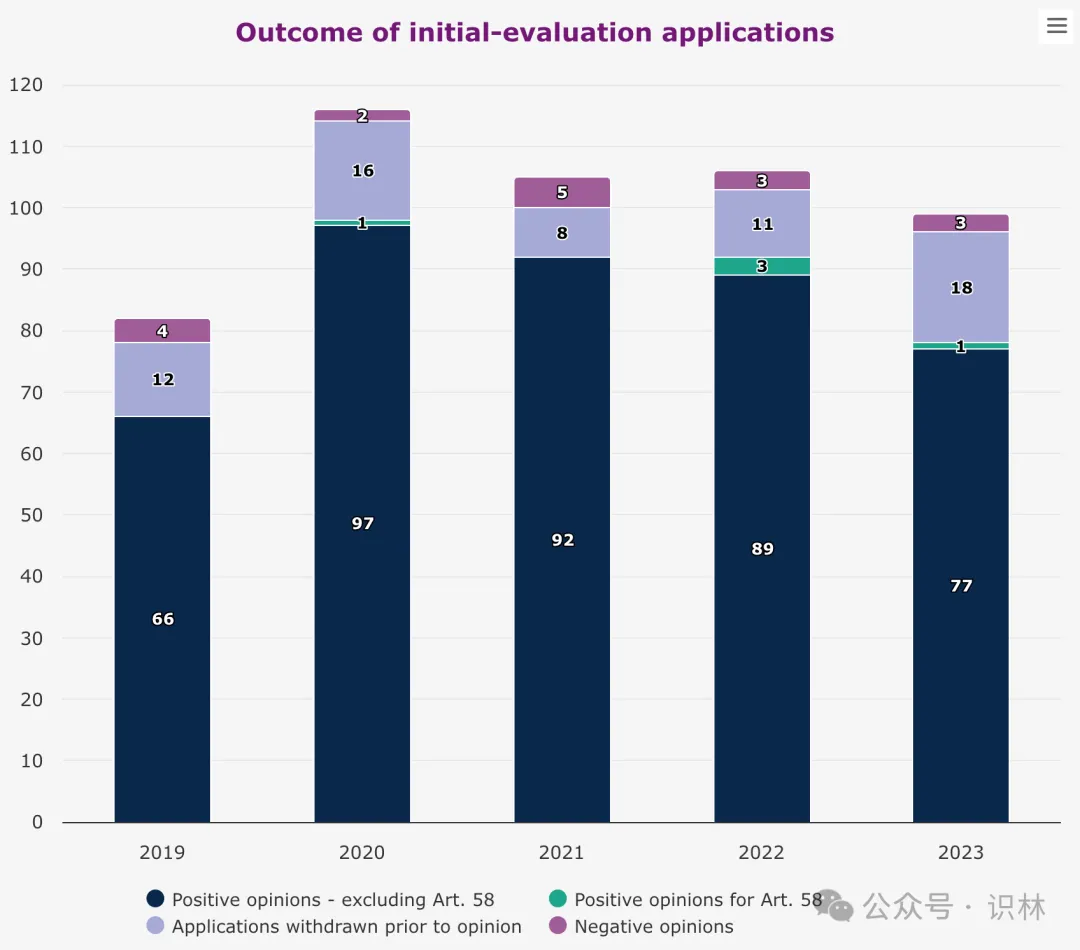
2023 年,EMA 建议批准了 77 个人用药,其中 39 个含有新活性物质 。许多药品代表了其治疗领域的重大进展。EMA 还推荐批准两个预防呼吸道合胞病毒(RSV)引起的下呼吸道疾病的疫苗 。EMA 还建议批准首个使用突破性基因编辑技术 CRISPR/Cas9 治疗两种罕见血液疾病(β 地中海贫血和严重镰状细胞病)。此外,EMA 还通过了两个关于供欧盟以外国家使用的药品的推荐意见。
2023 年建议批准的 77 个药中,3 个通过加速审评 (仅用于解决存在未满足医疗需求的药物)获得上市许可 建议,8 个药获得有条件上市许可建议(有条件许可允许在临床数据比通常要求的更少的情况下提前批准),1 个药是在特殊情况下获得许可建议(允许患者使用在标准许可下无法获批的药物)。17 个药获得孤儿药 认定,8 个药在获得上市许可建议之前失去孤儿药资格,即,仍被建议批准,但不再是孤儿药。另外,EMA 推荐扩展 77 项已获许可药物的治疗适应症。
兽药领域,EMA 推荐了 14 个药品上市许可,其中 9 个含有新活性物质,与 2022 年相比增加了三倍。其中 9 个为疫苗,包括 6 个新型生物技术疫苗。
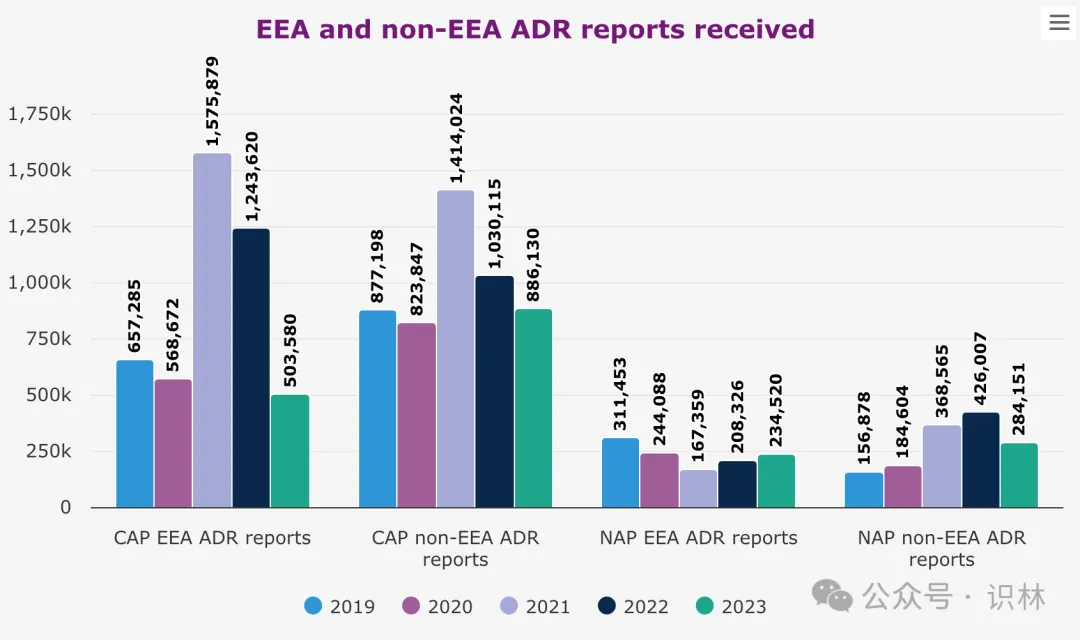
另外,报告中对药物不良反应(ADR)数据的报告显示出随着新冠疫情的缓解,EMA 的工作量已经恢复正常。去年,从欧洲经济区(EEA)发送的集中授权产品(CAP)不良反应 报告数量下降至约 50 万份,这一数字还不到 2022 年报告数量的一半,与疫情前的数据更加一致。
EMA 表示,疫情期间,包括患者报告在内的 ADR 报告率大幅上升,是大规模疫苗接种运动的结果,也是人们提高对报告任何疑似副作用重要性的认识的结果。
检查
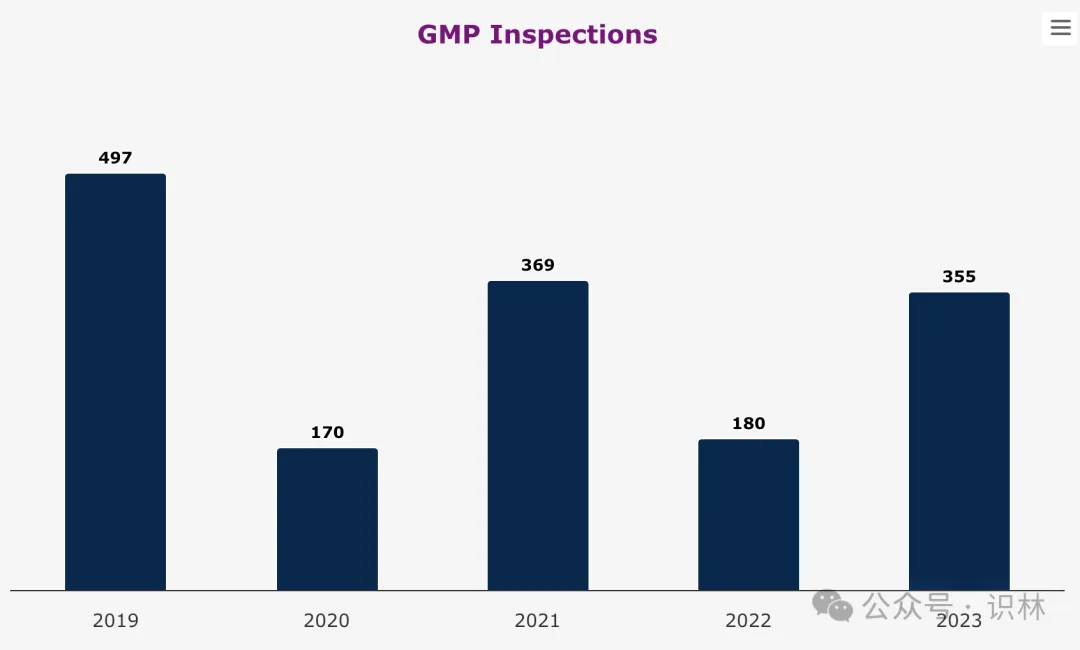
欧洲药品监管网络中,EMA 本身不执行检查,检查责任在各国主管机构,但 EMA 发挥重要协调作用。2023 年 EMA 要求的 GMP 检查数量上升至与 2021 年相当的水平。
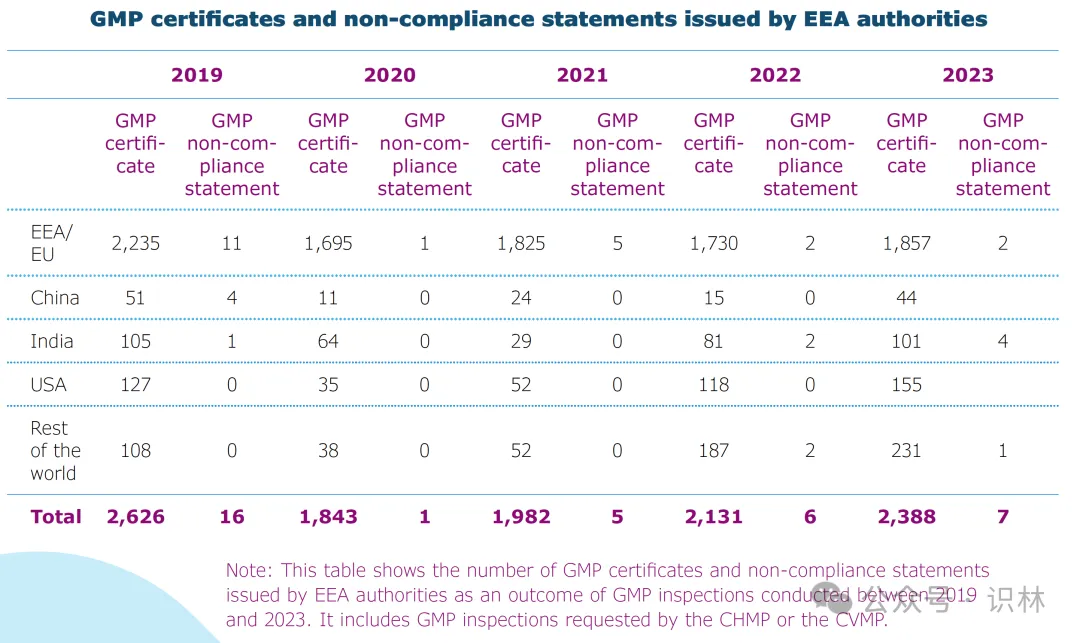
EEA 主管机构执行的 GMP 检查中有 7 次检查导致发出不合规声明。下图显示了 2019-2023 年间由各 EEA 主管机构执行的 GMP 检查之后发出的 GMP 证书和不合规声明情况。
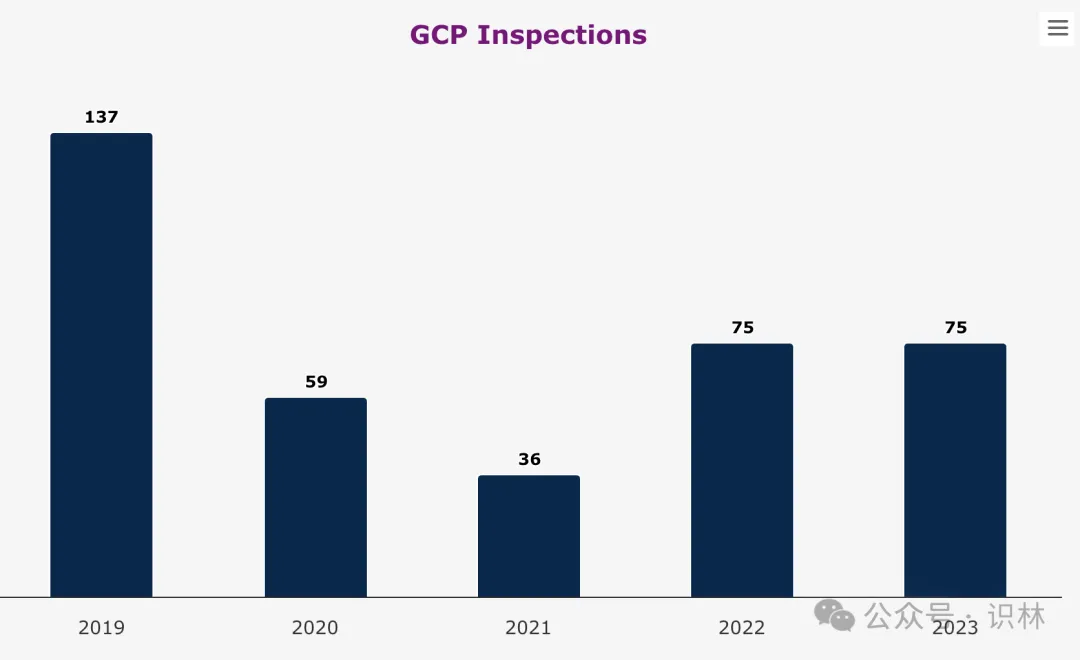
2023 年 EMA 要求进行的 GCP 检查为 75 次,与 2022 年持平,比 2021 年和 2020 年增多,但仍低于疫情前水平。
抗癌药探路者计划
2023 年,EMA 启动了一项名为“抗癌药探路者”的新计划,进一步支持可能对患者治疗产生重大影响的抗癌药的开发和批准。该计划探讨了 EMA 如何应用从新冠疫情中汲取的经验来改进整体药品审评。报告概述了“探路者”计划及其三大支柱的进展:加速对药品的审评、加强与利益相关者的对话,沟通获益和风险。
更多内容请阅览 2023 年报全文 。
作者:识林-椒
识林® 版权所有,未经许可不得转载
岗位必读建议:
QA :确保质量管理体系与EMA年度报告中的标准一致。注册 :关注EMA对注册流程的最新要求,确保注册策略的合规性。研发 :了解EMA对创新药物研发的指导,优化研发流程。文件适用范围:
文件要点总结:
监管合规性强调 :报告中强调了药品生产企业必须遵守EMA的监管要求,确保药品的质量和安全性。创新药物研发指导 :特别指出了对创新药物研发的支持和指导,鼓励企业进行创新。注册流程更新 :明确了注册流程中的变化,包括对申请材料和审批流程的更新。质量管理体系要求 :新增了对药品生产质量管理体系的具体要求,以提高药品的一致性和可靠性。市场准入策略调整 :报告中提到了对市场准入策略的调整,以适应不断变化的医药市场环境。以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。 QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。