首页
>
资讯
>
FDA首席副局长Woodcock撰文,谈美国临床试验多样化
出自识林
FDA首席副局长Woodcock撰文,谈美国临床试验多样化
2022-04-07
近日美国FDA首席副局长Woodcock在JAMA发布了一份研究报告,题目为“2015-2019年美国新药和生物制品 临床试验中的种族(Racial)和族群(Ethnic)代表性”,以了解美国临床试验中的受试者 在多大程度上代表了美国的人口多样性。
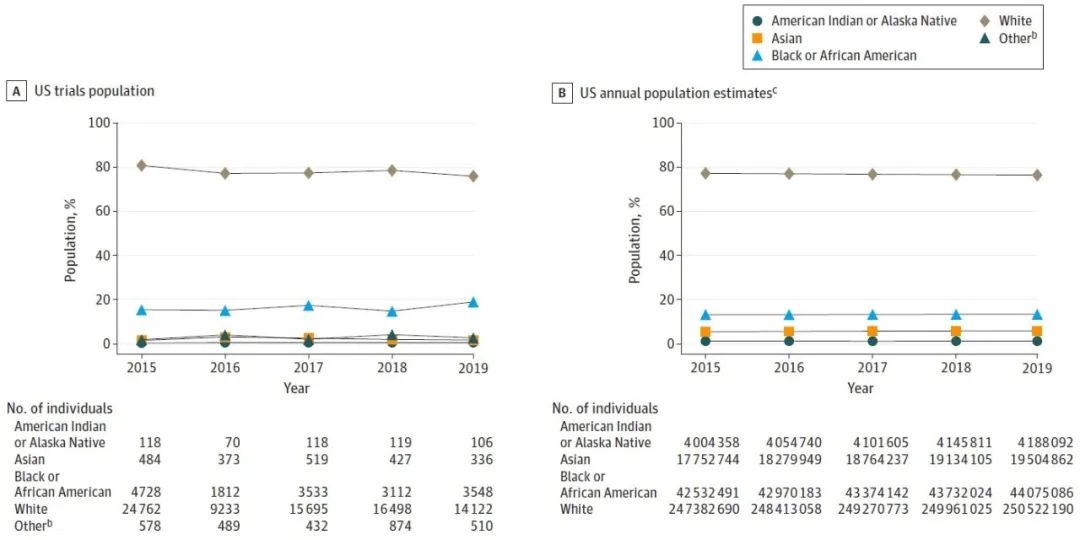
2015-2019年期间的517项临床试验[1] 中,来自美国试验场地的受试者102,596位,占全球受试者(292,766)的35%。从结果来看,在临床试验中,与人口普查数据相比,少数种族或族群的人口的代表性不足。黑人或非裔美国人在临床试验中的比例达到或高于美国人口普查水平,但其他少数种族或族群不足(表1和图1)。西班牙裔或拉丁裔的受试者比例从 10% 到 21% 不等(平均为15.3%,而人口普查为 18.5%),5 年中有 3 年的比例低于人口普查水平,大约 7% 到 13% 的族群数据丢失。
种族(Race)
临床试验比例范围(均值)
人口普查比例
黑人或非洲裔
15-19(16.3)
13.4
亚洲人
2-3(1.6)[2]
5.9
每周印第安人或阿拉斯加原住民
0.4-0.6(0.52)
1.3
白人
76-81(78.3)
76.3
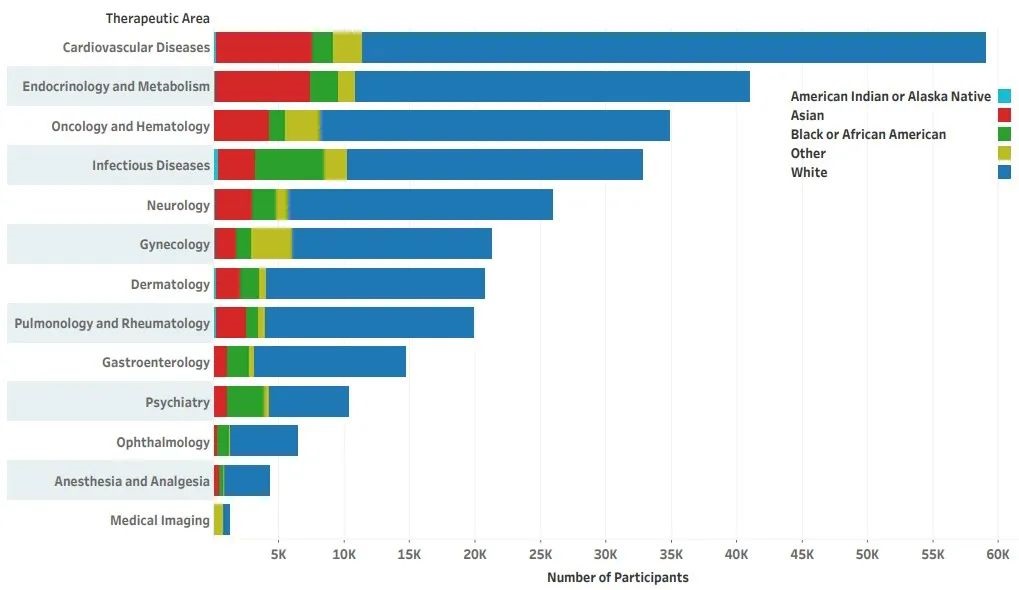
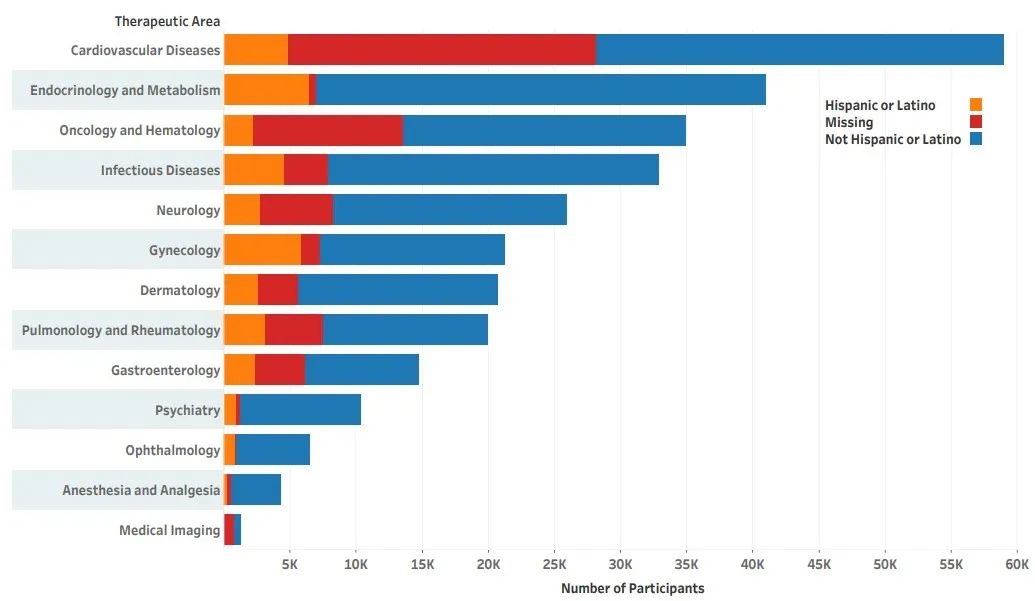
另外,在2020年FDA发布的《2015-2019 年药物试验快照摘要报告—临床试验参与和人口统计的五年总结和分析》 报告[3] 中,更为具体的展示了在不同疾病领域中,各种族或族群的受试者比例情况(图2和图3):
白人:临床试验参与比例最低的领域为精神疾病(51%),最高为心血管(90%)
黑人或非裔:参与比例最低的领域为医学成像(5%),最高为精神疾病(45%)
亚洲人:参与比例最低的领域为心血管试验(0.75%),最高为皮肤病(4%)
美洲印第安人和阿拉斯加原住民:参与比例最低的领域为医学成像(0.2%,最高为传染病(0.9% )
西班牙裔或拉丁裔:参与比例最低的领域为医学影像(1%),最高为传染病和胃肠病学(22% )
报告最后指出,直接比较试验和人口普查中的种族/族群比例并不是一个明确的评估,需要基于对疾病流行率和流行病学的情况,对多样性进行更详细的评估,但部分种族/族群的在临床试验中比例低还是凸显了增加美国临床试验受试者多样性的必要。
美国监管措施
根据21 CFR 314.50(d)(5)(v) ,FDA 期望申办者 招募的受试者反映了临床相关人群在年龄、性别、种族和族群方面的人口统计数据。《有效性的综合总结行业指南》 (Integrated Summary of Effectiveness)要求,在有效性综合总结中必须包括“按性别、年龄和种族亚组分析的有效性数据,识别任何特定亚组的给药的修改“的信息。亚群评价可以识别药物在亚群间的有效性特征的差异。如果发现这样的差异,则可能是重要的。亚组分析应被视为安全性和有效性的总体评价的一个组成部分,但是通常不应旨在支持特定亚组中数据的有统计意义的解释。
3月17日在众议院能源和商业委员会(House Energy and Commerce Committee)/健康小组委员会(Health Subcommittee)立法听证会上讨论的 22 项法案中有 4 项涉及或包含关于增加临床试验多样性的规定。
立法名称/编号
促进多样性的关键规定
多样化和公平参与临床试验 (DEPICT) 法案 (H.R. 6584)
FDA 发布法规,要求申办者为临床试验制定多样性计划,包括:按人口统计亚组估计的病症或疾病的患病率;按年龄、性别、种族和民族划分的入组目标;入组的理由;以及如何实现目标的行动计划。
申请人必须提交一份年度报告 ,说明实现注册目标的进展情况以及需要对多样性行动计划进行的任何更新。
由于人口统计数据不足,FDA 可能会要求进行批准后研究或上市后监督,尽管该机构也可以放弃这些研究。
FDA 收集利益相关者对 COVID-19大流行期间制定的临床试验灵活性的意见,以及将某些试验中断缓解策略纳入当前或新指南的建议,以改善临床试验的访问和不同人群的注册。
要求 FDA 发布关于增加临床试验和研究注册多样性的进展的年度报告。
通过公平研究(DIVERSE)试验法案(H.R. 5030)使研究多样化
FDA 发布关于开展具有有意义的人口多样性的分散临床试验(decentralized clinical trials)的指南草案,包括患者参与、登记和受试者的种族、民族、年龄、性别和地理多样性。指南应说明如何适当使用数字健康技术或其他远程评估选项,例如远程医疗。
为药物或设备制造商向临床试验受试者免费提供数字健康技术提供安全港。
为临床试验受试者 提供薪酬安全港,促进公平纳入所有相关人口和社会经济人群的患者,并与试验参与有关。
神经科学公平和阿尔茨海默症临床试验 (ENACT) 法案 (H.R.3085)
美国国立卫生研究院通过合作协议、赠款、培训奖励和在少数族裔高度集中的地区建立临床试验场地,提供激励措施和外展服务,以增加阿尔茨海默病研究的多样性。
治愈 2.0 法案 (H.R. 6000)
要求政府问责办公室报告审查卫生和公共服务部如何解决代表性不足的人群参与临床试验的障碍以及解决这些障碍的建议。
要求 HHS 制定公众意识运动,以提高对临床试验的认识和理解,特别是在少数族裔社区。
需要召集一个常设工作组以使 ClinicalTrials.gov 更加用户友好。
美国立法者表示强烈支持增加FDA用户收费立法条款,以增加临床试验的多样性,但这些措施是否会对申请人施加可执行的义务,或者仅仅激励更多样化的注册,还有待观察。
Moderna的mRNA疫苗在临床试验多样化中的实践
为了提高透明度,Moderna在进行mRNA疫苗 III期临床试验(COVE)期间就公开了试验方案 。另外,为了临床试验的多样性,Moderna停止招募白人受试者参与试验,转而专注非白人群体的招募。
停止招募白人受试者的决定源于早期的预测,即 2020 年 7 月启动的试验将代表一个不代表美国整体人口的人群。Ivarsson 指出,直到 2019 年,美国的临床试验中约有 94% 是白人,Ivarsson表示
“我们花了一两个星期的时间来实施,因为......我们希望非常尊重那些已经来到试验场地并被安排进行检查的人。因此,我们允许那些不分种族/族群的人前来参观,一旦完成,我们要求这些场地向非白种人提供所有剩余的名额。”
最终,根据 2021 年 2 月 4 日发表在《新英格兰医学杂志》上的结果,COVE 的 30,351 名受试者 , 79.2% 白人、20.5% 拉丁裔、10.2% 黑人、4.6%亚洲人、0.8% 美洲原住民或阿拉斯加原住民,0.2 % 太平洋岛民和 2.1% 多种族;20.5% 是任何种族背景的西班牙裔或拉丁裔。根据人口普查数据,这些群体的细分总体上基本与他们在美国整体人口中所占份额相符。由于拉丁裔具有多种种族身份,总数加起来超过 100%。
参考文献
【1】Milena Lolic,Richardae Araojo,Melvyn Okeke,JanetWoodcock. Racial and Ethnic Representation in US ClinicalTrials of New Drugs and Biologics, 2015-2019. JAMA. 2021 Dec 7;326(21):2201-2203. doi: 10.1001/jama.2021.16680.
【2】Sue Sutter.Clinical Trial Diversity Measures Primed For Inclusion In US FDA User FeeLegislation. Pinksheet. 2022.03.17
【3】Alaric DeArment.How Little Moderna Made Big Waves In Trial Diversity. Scrip. 2022.02.07
【4】FDA. 2015-2019 DRUGTRIALS SNAPSHOTS SUMMARY REPORT—Five-Year Summary andAnalysis of Clinical Trial Participation and Demographics. 2020.11
[1] 2015 年至 2019 年间发表的新分子实体 和原研生物制品 关键临床试验的汇总。
[2]原文“The proportion of Asian participantsranged from 2% to 3% (mean, 1.6% vs Census, 5.9%)”
[3] 2015 年,FDA启动了药物试验快照计划,这是一项透明倡议,强调支持 FDA 批准新分子实体或原研生物制品的关键临床试验中的人口统计数据。
作者:识林-木兰
识林® 版权所有,未经许可不得转载
适用岗位:
注册(RA) :必读。负责理解ISE的格式和内容要求,以确保新药申请(NDA)或生物制品许可申请(BLA)符合FDA的指导原则。临床(Clin) :必读。需要根据ISE的要求设计和执行临床试验,以及分析临床数据。研发(R&D) :必读。在药物开发过程中,需要参考ISE来整合和分析药物的有效性数据。工作建议:
注册(RA) :确保ISE包含所有必要的部分,如个别研究的列表和简要结果、研究设计的分析、整体有效性结果的分析等。 与FDA沟通,讨论替代方法的可能性,以满足适用法规的要求。 临床(Clin) :在设计临床试验时,考虑ISE中提到的各种分析,如亚组分析、剂量反应分析等。 确保临床试验数据能够支持ISE中要求的各种分析。 研发(R&D) :在药物研发过程中,整合ISE的要求,确保研究结果能够全面反映药物的效益。 关注ISE中提到的研究设计和执行的关键要素,以提高研究质量和数据的可靠性。 适用范围:
要点总结:
ISE内容要求 :强调了ISE应包含的内容,如个别研究的列表和简要结果、研究设计的分析、整体有效性结果的分析等。亚组分析 :明确了对亚组分析的要求,包括基于人口统计学特征和其他相关因素的亚组。剂量反应分析 :强调了对剂量反应或血药浓度反应关系的分析,以支持剂量推荐。时间效应分析 :要求展示药物效果的时间过程,包括效果的持续性和耐受性。探索性研究 :鼓励报告基于未在协议中指定的终点、患者亚组和汇总数据的探索性分析结果。以上仅为部分要点,请阅读原文,深入理解监管要求。