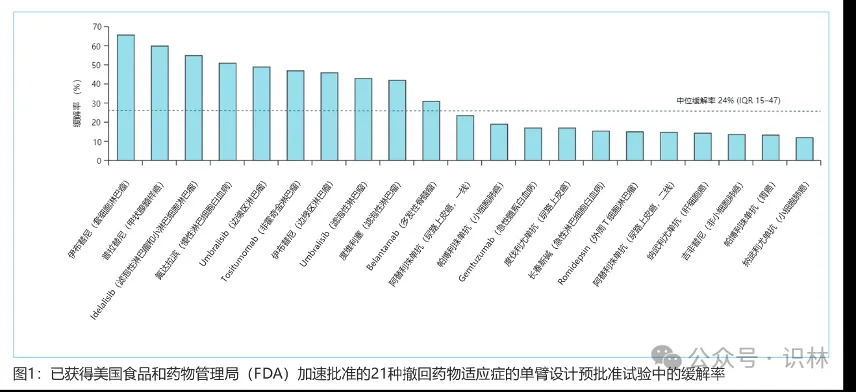
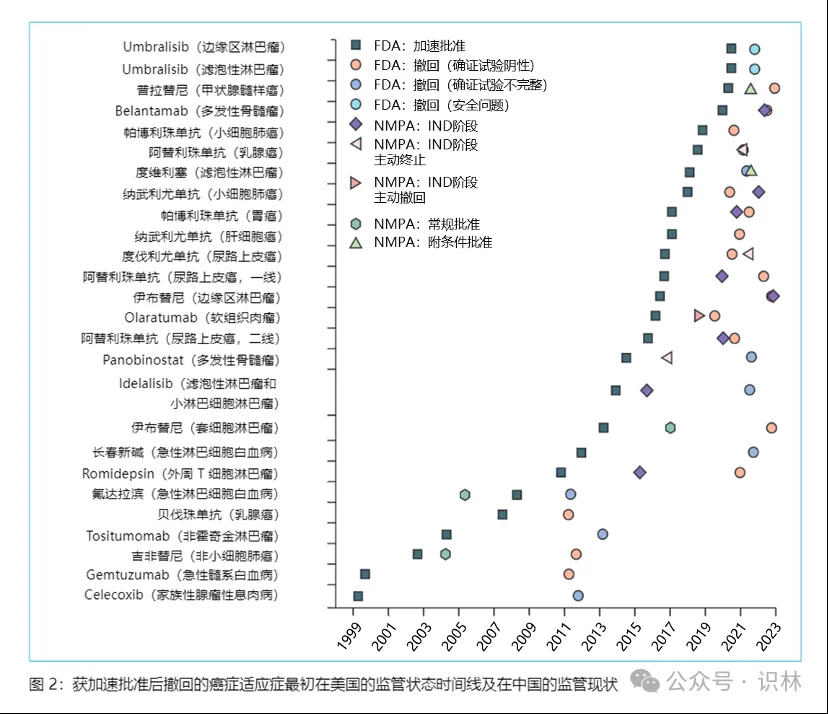
大多数在美国获得 FDA 加速批准后被撤市的肿瘤适应症的确证性临床试验结果为阴性,所有这些阴性确证性临床试验均为随机对照试验,并将总生存期作为主要或次要疗效终点。广泛用于预测总体生存获益的肿瘤替代终点一直存在争议。替代终点有时不能转化为肿瘤患者总生存期获益的原因复杂且众多,包括严重的药物毒性反应,允许对照组患者交叉到实验组,以及由于研究并不是围绕总生存期作为主要疗效终点而设计导致潜在的统计功效(statistical power)不足。因此,仅通过判断替代终点的改善来预测总体生存获益变得越来越具有挑战性。尽管 FDA 于 2018 年首次发布了一份表格,列出了其用作加速批准程序的替代终点,但这些替代终点与总生存期的相关程度并未公布,同样有研究表明中国附条件批准的抗肿瘤药物的替代终点与总生存期之间缺乏既定证据。这些都强调了全球合作建立替代终点与总生存期之间相关性数据库的重要性。
以往研究显示欧洲药品监督管理局(EMA)共批准了9个(39%)美国FDA已撤市的加速批准肿瘤适应症,这个比例要明显多于我国批准的数量(5个, 19%),这也提示不同监管机构对加速批准肿瘤药审评方面存在差异。研究者指出,由于制药公司通常先在美国提交上市申请,因此抗肿瘤药物在中国上市会出现延迟,加强机构之间的合作和数据共享(例如,通过FDA 建立的 Orbis 项目)将有助于减少差异,特别是对于通过加速批准途径上市的抗肿瘤药物。建议我国可要求针对加速批准/附条件批准肿瘤药物应及时提交最新的有效性和安全性证据,尤其是因国外确证性临床试验失败或未能及时完成导致在其他国家撤市的品种,并重新进行获益-风险分析,以确保患者真正的临床获益。2023年8月,我国发布《药品附条件批准上市申请审评审批工作程序(试行)(修订稿征求意见稿)》,提出在提交附条件批准新药上市申请前应启动确证性临床试验并入组首例患者,这些监管调整预计将减少附条件批准药品临床获益的不确定性。