|
首页
>
资讯
>
FDA 发布 2021 年药品指南制定计划
出自识林
2021-01-29
美国 FDA 药品审评与研究中心(CDER)于 1 月 25 日发布了其 2021 年药品方面的指南制定计划,涵盖 18 个类别,共 105 篇计划新增或修订的指南,比去年 2020 年计划的 88 篇文件增加了 18%,其中 42 篇是 2021 年的新指南,剩余的则是从 2020 年的计划清单中遗留下来的。和去年一样,计划发布的指南大部分涉及仿制药、药品质量/CMC 和程序性工作,不过仿制药和程序性工作两个类别中保留了大部分上年度甚至是 2019 年度未如期发布的指南。
实质性证据标准指南
临床/医学类别列出了 13 篇指南,其中只有 4 篇是从 2020 年计划中顺延下来的,另外 9 篇是新出现在这份清单上的。其中值得注意的一篇指南是《满足基于一个充分且良好对照的临床研究以及确证性证据的实质性证据标准》指南。该计划发布的指南标题是 2019 年 12 月发布的一份指南草案《证明人用药品和生物制品的有效性的实质性证据》中的第 IV.B 小节的标题。【FDA 就证明药品有效性所需的实质性证据提供更多灵活性 2019/12/23】
在 2019 年这份指南草案的该小节中,FDA 介绍了在确定依赖单个充分且良好对照的临床研究加上确证性证据是否合适时应考虑的因素。指南草案进一步解释了确证性证据可能包括:相关疾病领域的研究,真实世界证据,机理证据以及疾病自然史。2019 年的指南中包括四个例子,证明一个单独的充分且良好对照的研究何时可以建立有效性证据:
- 对已获批药品的新适应症进行一项充分且良好对照的临床研究 , 并辅以已证明药物对其它紧密相关的获批适应症的有效性的现有充分且良好对照的临床研究;
- 一项充分且良好对照的临床研究,并提供强有力的机制支持的数据;
- 一项充分且良好对照的结果令人信服的临床研究 , 并有该疾病自然史的其它数据支持;
- 一项关于新药的充分且良好对照的临床研究,以及有关相同药理学类别其它药物有效性的科学知识的支持。
在罕见病背景下,经常会出现有关通过一个充分且良好对照的试验来证明实质性证据是否可行的问题,因为罕见病的特性,可能难以进行多个随机试验。
真实世界证据
CDER 计划在 2021 年制定三篇真实世界证据相关的指南文件:
- 真实世界数据:评价电子健康记录和医疗索赔数据以支持药品和生物制品的监管决策
- 使用真实世界数据和真实世界证据来支持药品和生物制品监管决策的监管考量
- 使用登记(registries)作为真实世界数据源用于 FDA 申报
根据《21 世纪医药法案》,FDA 在 2018 年 12 月发布了真实世界证据框架,但直到 2021 年 12 月才会发布有关真实世界证据做出监管决策的指南草案。【FDA 对在真实世界证据框架中使用观察性研究犹豫不决 2018/12/21】到目前为止,FDA 仅发布了一篇针对药品和生物制品的真实世界数据/证据指南草案,指南解释了申办人应如何确定将此类证据纳入产品申请中。【真实世界证据支持申请批准的时代已经到来?2019/05/20】
在昨日的资讯中,我们介绍了 FDA 首席副局长 Amy Abernethy 表示,新冠疫情正迫使 FDA 和药品开发者更快地适应真实世界数据集。【疫情加速 FDA 对真实世界数据的使用 2021/01/28】
个体化治疗,生物类似药
2021 年的指南制定计划还包括针对严重衰弱或危及生命的疾病的个体化反义寡核苷酸药物产品的三篇指南,计划发布的这三篇指南将解决:
- 支持研究性新药申请申报的临床建议;
- 化学、生产和控制方面的考量(也出现在 2020 年的指南计划中);
- 非临床测试。
本月初,CDER 发布了一篇关于个体化反义寡核苷酸药物研究用新药(IND)申请的程序建议指南。【FDA 发布个体化反义寡核苷酸药物申报的指南草案 2021/01/05】
2021 年的指南计划清单中还列出了与生物类似药有关的四篇指南,其中包括首个可互换生物制品专营权的指南,以及针对开发生物类似药和可互换生物产品的具体产品类别的建议。FDA 还计划制定有关生物参照产品和生物类似产品的促销标签和广告注意事项的问答文件,以及对 2018 年 7 月生物类似药产品标签定稿指南的修订。
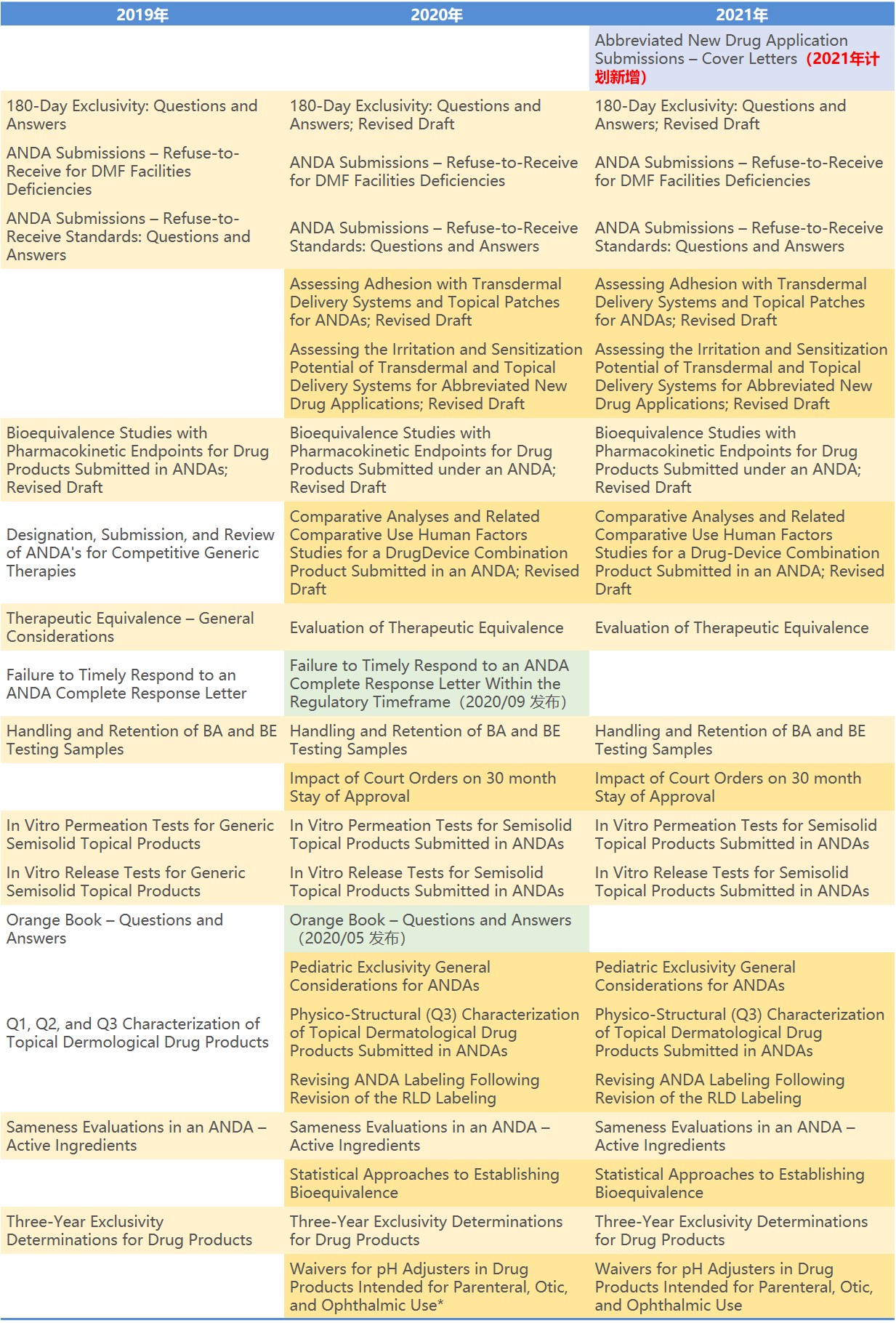
仿制药
2021 年指南计划清单中列出的指南数量最多的是仿制药类别,共计划发布 20 篇指南,但是其中仅有一篇是 2021 年计划新增的,而另外 19 篇是在 2020 年指南计划上但未如期发布的指南。通过下表可以一目了然的看出哪些指南拖延了多年仍未发出,这些未如期发布的指南中相当多是业界极为关注的内容。
作者:识林-椒
识林®版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
岗位必读建议: - 注册部门:深入了解FDA关于证明药品和生物制品有效性的指导草案,以便在新药申请(NDAs)和生物制品许可申请(BLAs)中准确呈现有效性证据。
- 研发部门:掌握临床试验设计、终点选择和统计考量,确保临床研究的质量和结果的科学性。
- 临床部门:熟悉临床证据的质量要求,包括试验设计、终点和统计分析,以指导临床试验的规划和执行。
文件适用范围:
本文适用于化学药和生物制品,包括创新药和仿制药,由美国食品药品监督管理局(FDA)发布,主要针对大型药企、Biotech公司、跨国药企以及CRO和CDMO等企业。 文件要点总结: 实质性证据标准:强调药品和生物制品必须通过“实质性证据”来证明其有效性,这通常需要充分和受控的临床研究。 临床证据的质量:讨论了试验设计、试验终点和统计考量对临床证据质量的影响,以及如何通过这些因素来确定有效性。 临床证据的数量:详细说明了基于两次充分和受控的临床研究、一次加上确认性证据或依赖于FDA之前对已批准药品有效性发现的证据来满足实质性证据标准的情形。 特殊情况下的灵活性:在疾病危及生命或严重致残且医疗需求未得到满足、疾病罕见或进行人体有效性试验不道德或不可行的情况下,可能需要额外的灵活性。 安全性与有效性的综合考量:虽然实质性证据的发现对FDA批准至关重要,但批准决定还需要确定药品对预期使用安全,这涉及到对药品风险和益处的综合评估。
以上仅为部分要点,请阅读原文,深入理解监管要求。 适用岗位: - RA(注册专员):必读。需理解生物类似药的标签要求,以便在提交351(k)申请时,确保标签信息符合FDA规定。
- QA(质量保证专员):必读。需确保标签内容的准确性和合规性,以及在产品生命周期内对标签的更新和维护。
- 研发:必读。在开发生物类似药时,需了解标签中应包含的数据和信息,以支持产品的安全性、纯度和效力。
- 市场:必读。需了解生物类似药的标签信息,以确保市场推广材料和沟通符合FDA的标签要求。
工作建议: - RA:在准备和提交生物类似药的标签时,应与FDA沟通,确保所有要求得到满足,并在产品获批后持续监控标签的合规性。
- QA:监督标签的准确性和更新,确保所有变更都符合FDA的规定,并及时更新。
- 研发:在临床研究和分析研究中,应收集和分析数据,以支持标签中的声明,并确保这些数据反映在标签中。
- 市场:在推广材料和沟通中,确保所有信息与FDA批准的标签保持一致,避免误导。
适用范围:
本文适用于生物制品中的生物类似药,包括创新生物类似药和仿制生物类似药,由美国FDA发布,适用于Biotech、大型药企、跨国药企以及CRO和CDMO等企业类别。 要点总结:
FDA的指南强调生物类似药的标签开发应基于与参考产品的高度相似性,而非独立证明其安全性和有效性。标签应包含参考产品标签中的相关数据和信息,并进行适当修改。生物类似药的标签必须满足医师标签规则(PLR)和妊娠与哺乳标签最终规则(PLLR)的要求。产品标识应根据信息呈现的上下文进行,可能包括生物类似药名称、参考产品名称或核心名称。标签内容的呈现应针对生物类似药寻求的适应症和剂量方案,并且与参考产品先前批准的语言保持一致。如果生物类似药申请者获得了少于参考产品所有适应症的许可,则某些参考产品标签中的文本通常不会被包括在生物类似药标签中。标签修订应反映新的安全信息,并在产品生命周期内更新。对于寻求额外适应症许可的情况,生物类似药申请者可以通过提交351(k)申请的前置批准补充文件来实现。 以上仅为部分要点,请阅读原文,深入理解监管要求。 |