首页
>
资讯
>
FDA 四款获批基因治疗产品及高效开发经验
出自识林
2020-06-16
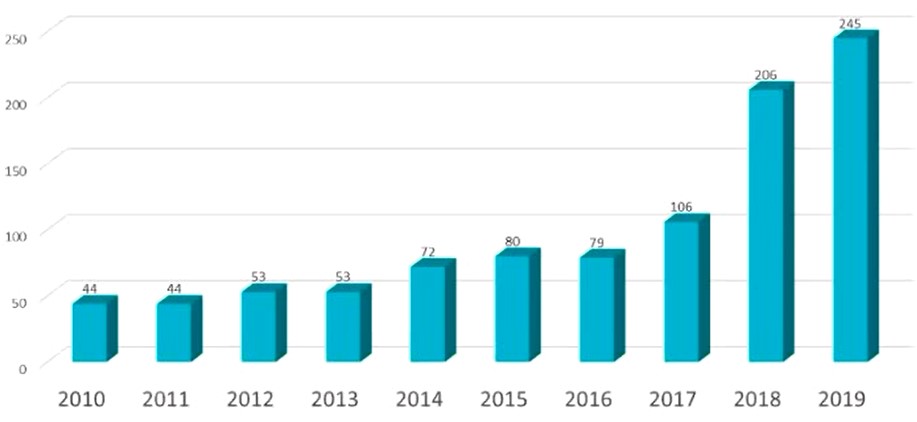
基因治疗的发展得益于科学技术进步,包括2003年10月完成的人类基因组计划(99%的人类基因测序,精确率达99%)、新载体的开发(包括腺相关病毒和慢病毒等)以及基因编辑技术等。图1为2010-2019间提交到FDA的基因治疗产品IND数量,显示了基因治疗产品开发的迅猛发展,2019年共有245项基因治疗产品IND 提交到FDA,是2016年的3倍多。
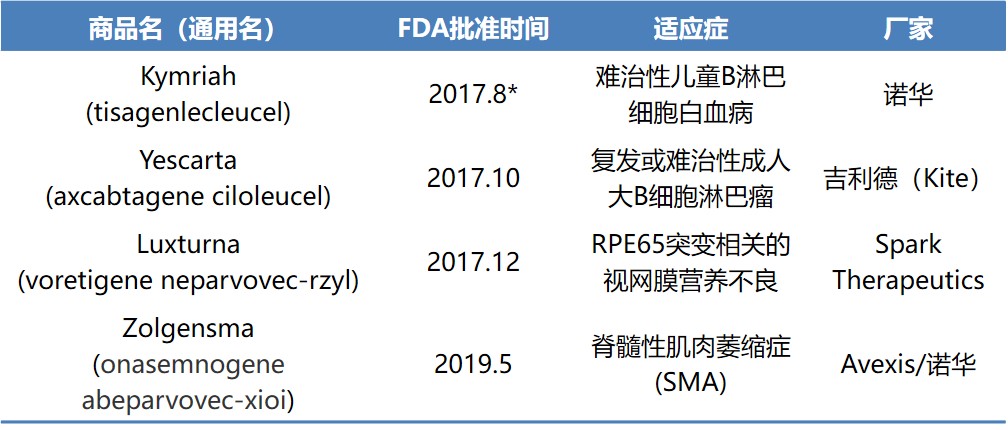
目前,FDA共批准了4款基因治疗产品(表1)。6 月 1 日举行的 ISPE 生物药制造网络研讨会上,美国FDA生物制品审评与研究中心(CBER)组织和先进疗法办公室(OTAT)主任 Wilson Bryan重点介绍了其中两款产品 — Luxturna和Zolgensma,以及如何进行基因治疗产品的高效开发。
*Kymriah 2018年5月被批准用于复发或难治性成人大B细胞淋巴瘤二线以上治疗
Luxturna是一款首创(First in class)基因治疗 产品,适用于与RPE65突变相关的视网膜营养不良患者的治疗。疾病临床表现为婴儿期失明或者患有夜盲症、渐进性视野丧失。所有患者最终都将完全失明。这是一种罕见病,美国每年有1000-2000名患者。此前没有获批治疗药物。
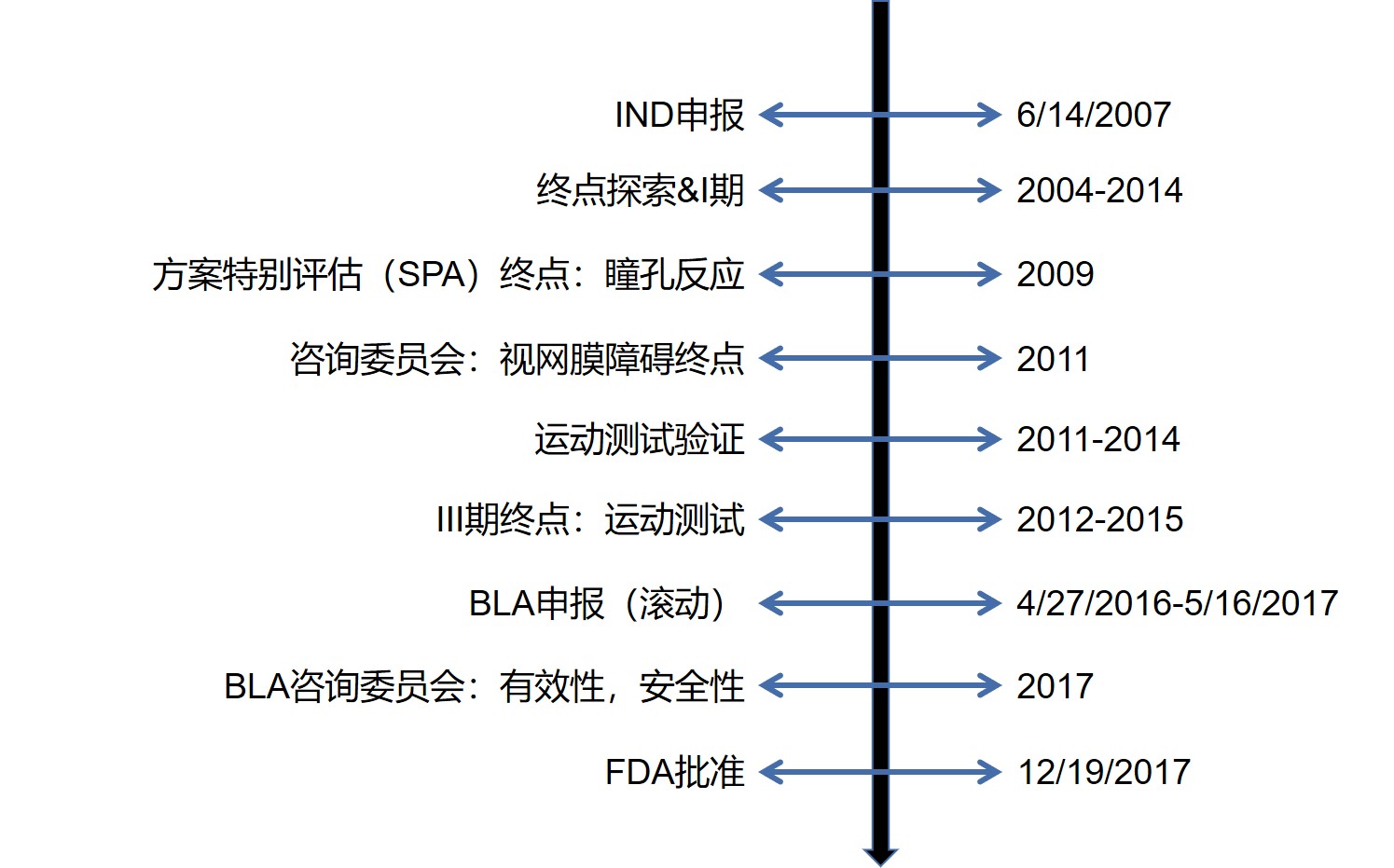
Luxturna的研发时间轴如图2,从IND申报到获批上市用了超过10年时间。值得注意的是,在IND提交3年之前即开始了终点探索,共进行了10年。Wilson强调:在研发早期,远早于IND提交时就应考虑什么临床终点 会是有效的,临床终点的确定可能花费很长时间。
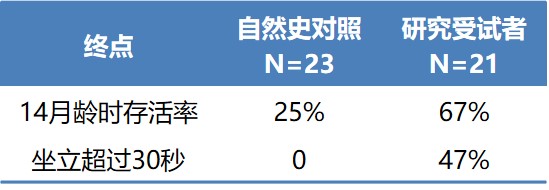
Zolgensma的适应症为脊髓性肌肉萎缩症(SMA)。1型SMA是重度的SMA,6月龄前发病。通常表现为吸乳能力差、吞咽困难、呼吸衰竭、无法独坐。发病率为万分之一。Zolgensma的III期临床试验结果如表2。Wilson强调:本实验并未设置对照组,使用的是自然史对照,而且这项自然史研究的开展时间比Zolgensma研究早了超过10年。FDA允许在某些条件下使用自然史对照。另外,Zolgensma的III期临床试验入组受试者为21人,I期和III期入组受试者合计少于40人。
Luxturna和Zolgensma经验小结:
1.治疗罕见病的产品可以基于较少数量的患者研究而获得FDA的批准。
高效开发建议
那么,如何进行基因治疗产品的高效开发?Wilson从以下3个角度给出建议:
1. 准备
这包括基础科学家和临床医生之间应尽早合作;临床前研究开始时,即应起草临床研究设计;设计并进行自然史研究,以支持后续的药物开发(这需要很长时间)。
2. 团队合作
需要科学家、学术研究者、申办者 、基金组织、患者、患者权益团体和监管机构之间通力合作。
3. 试图全胜
Wilson将临床试验比作棒球比赛,全垒打(hit a home run)即一击将球击出场外大获全胜。他认为基因治疗产品的临床试验没有必要按照I期、II期、III期的刻板顺序进行临床试验,可以朝着全垒打的方向努力,I期试验同时可以是III期试验。他指出,可以在设计首个人体临床试验时提供有效性证据(如,包括随机对照)。另外,应该在首个人体临床试验前尽可能解决生产问题,而不要等到II期、III期再进行生产变更 。因为他见过许多基因治疗产品具有临床有效性证据,但由于生产问题,不得不等待一年或几年之后才得以获批,这着实让人感到可惜。
作者:识林-禾® 版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。