|
首页
>
资讯
>
FDA 2021 仿制药论坛上总结的政策和工作重点
出自识林
FDA 2021 仿制药论坛上总结的政策和工作重点
2021-05-04
美国 FDA 一年一度的仿制药论坛于 4 月 28 至 29 日召开,会上讨论了很多重要话题。今天我们先来看看整体有关仿制药审评和政策方面的内容。
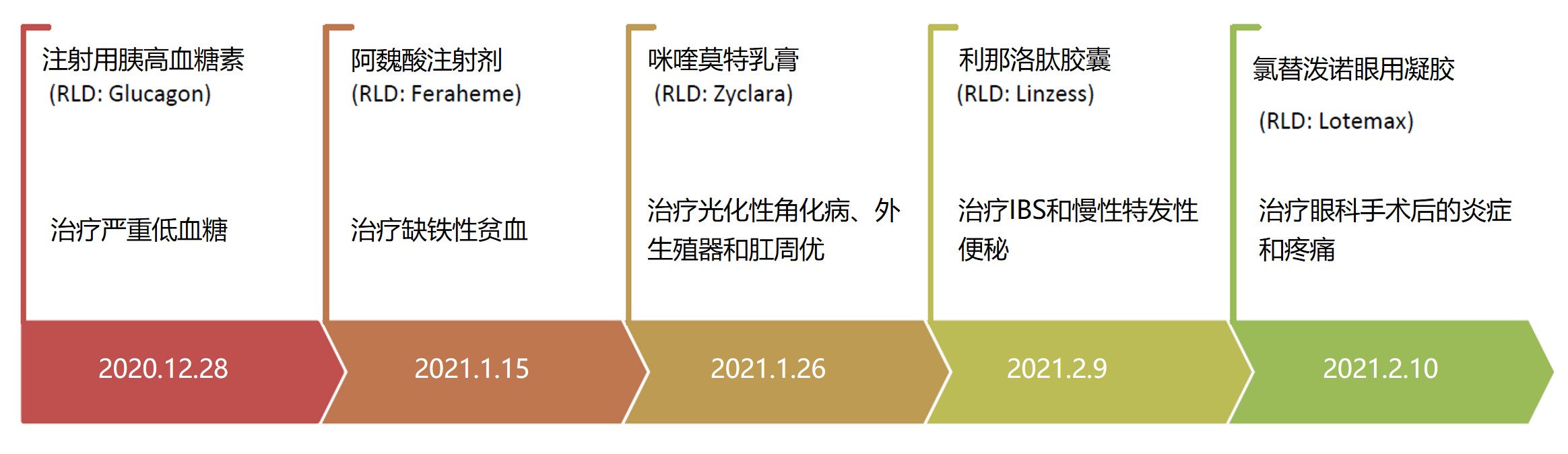
首先仿制药办公室(OGD)主任 Sally Choe 做了有关 OGD 整体工作的报告。Choe 指出,为满足与新冠大流行相关的需求,OGD 在 2021 财年优先审评了 50 件 ANDA 和 800 多件补充申请。Choe 还列举了 2021 财年到目前为止值得关注的首仿批准:
她指出,仅在 2020 财年,就有 32 篇新的或修订的具体产品指南(PSG)引入了一种体外方法来证明生物等效性。此类方法提供了较低的监管障碍,同时确保了仿制药产品具有生物等效性。
自 1984 年 Hatch-Waxman 法案颁布以来,随着更新和更独特产品的开发和批准,药品的复杂性也在不断增加。随着药品复杂性从简单的小分子发展到具有复杂活性成分、剂型、配方或给药途径的产品以及作为药械组合的产品,复杂药品带来了新的挑战性问题。对这些复杂产品的审评需要对属性进行特殊评估,而在 Hatch-Waxman 法案颁布之初的小分子固体口服制剂中并没有这些属性。
FDA 已与包括大学和研究组织在内的外部机构签订合同并建立合作关系,这类合作以及 FDA 内部专家组的工作使得许多复杂产品的批准成为可能。2021 财年 OGD 批准的一些值得关注的复杂仿制药如下:
产品质量办公室的科学副主任 Larry Lee 介绍了其办公室在 2020 年的一些行动要点。包括使用替代工具避免了 153 次批准前检查;293 项加速质量评价等等,在药品质量办公室 2020 年度报告中有详细说明。【全文翻译FDA 药品质量办公室 2020 年度报告 2021/02/15】
质量政策重点
药品质量政策办公室 Ashley Boam 在会上介绍了 OPQ 和 OGD 面临的各种政策问题,并概述了 2021 财年的质量政策重点:
- ANDA:原料药和制剂的稳定性检测问答指南
- NDA、ANDA和 BLA 的原料药和制剂的质量和稳定性测试以及制剂的相关标签声明指南
- 注射产品中的可见颗粒检查指南
- 通过肠内管饲给药的药品:体外测试和标签建议指南
- 非无菌药品生产中的微生物质量考量指南
- CDER 自愿性共识标准认可计划(定稿)
- 拟议规定:已获批申请的批准后变更
- 内部:由于旅行限制而无法完成检查时使用的与“替代工具”相关的实施活动
- 质量管理成熟度
- 国际协调
她还讨论了《联邦食品、药品和化妆品法案》(FDCA)第 510(j)(3) 小节,该小节增加了一项要求:根据第 510 节注册药品场地的人员必须每年报告为商业销售生产的每种上市药品的数量。此外,她还概述了《冠状病毒援助、救济和经济安全法案》(CARES 法案)对风险管理计划的新要求以及新冠应对期间发布的各种指南。
数据监督
药品质量办公室质量监督办公室代理主任 Jennifer Maguire 讨论了其办公室审查的监督活动。她的办公室不采取执法行动,而是评价可供 FDA 使用的各种数据集。如果所审查的数据中可能存在问题,监督办公室会将其提交给合规办公室。监督的数据类型包括但不限于:
- 质量管理成熟度(未来)
- 外部数据
- 国外监管机构信息
- 公共信息 — 社交媒体、消费者评论、博客、新闻媒体等。
关于仿制药论坛的更多内容,识林将会在本周继续分主题报道。
识林®版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
必读岗位及工作建议: - QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。
- QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。
- 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。
- 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。
适用范围:
本文适用于化学药领域的原料药生产,包括创新药和仿制药,适用于大型药企、跨国药企以及CRO和CDMO等企业类别,发布机构为国际通用标准。 文件要点总结:
原料药的生产质量管理规范强调了从质量管理到生产控制的全过程管理。首先,文件明确了质量管理的原则和机构职责,特别强调了质量保证和质量控制的重要性,并规定了自检、产品质量回顾以及质量风险管理的具体要求。在人员方面,规定了资质、培训和卫生要求,确保员工符合岗位需求。厂房与设施章节详细规定了设计建造、公用设施和特殊隔离要求,以保证生产环境的适宜性。设备章节则涉及设计建造、维护保养、校准和计算机化系统的要求,确保设备运行的可靠性。文件还特别提到了无菌原料药的生产特点,包括生产工艺、厂房设施设备设计、生产过程管理以及环境控制等,这些都是确保原料药质量的关键环节。 以上仅为部分要点,请阅读原文,深入理解监管要求。 岗位必读建议: - QA:确保口服固体制剂的生产过程符合本指南要求。
- 生产:遵循生产管理章节的指导,确保生产过程的合规性。
- 研发:在设计和选型设备时,参考本指南以确保设备符合生产需求。
- 临床:在产品实现和验证阶段,确保临床试验用药品的质量符合要求。
文件适用范围:
本文适用于口服固体制剂的化学药品,包括创新药和仿制药。适用于中国药企,包括大型药企、Biotech以及CRO和CDMO等。 文件要点总结: - 质量风险管理:强调了质量风险管理在口服固体制剂生产中的重要性,包括原则和工具的应用。
- 生产管理:明确了生产过程中的关键控制项目,如批次管理和清场管理。
- 设备要求:规定了生产设备的设计、选型、校验、清洗、维护和使用记录。
- 生产过程控制:概述了工艺设计和过程单元操作的详细要求,包括配料、粉碎、混合等。
- 物料管理:强调了物料的接收、储存、分发、退库以及检验与放行的管理。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |