|
首页
>
资讯
>
从教训开始学习 – 看看回复483有哪些常犯的错
出自识林
2019-08-06
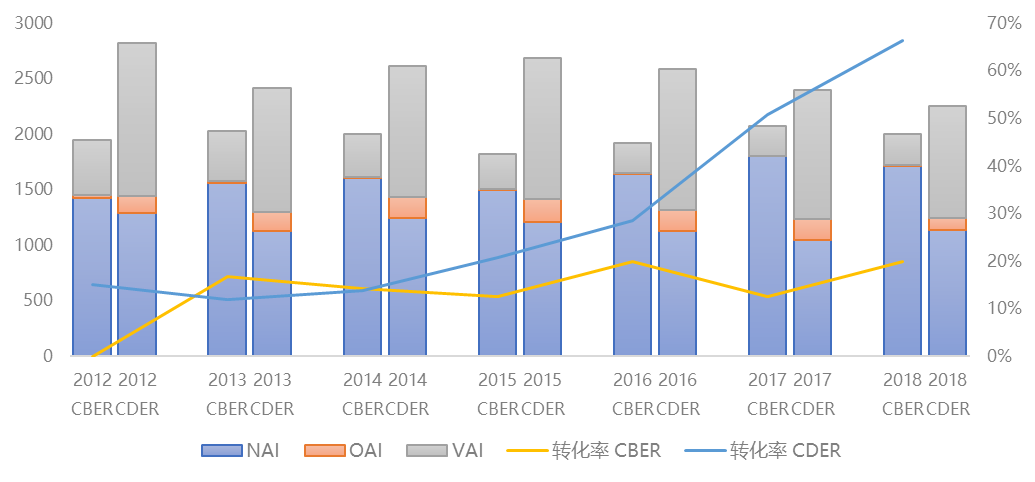
是不是检查结果不好就会收到警告信呢?根据识林483数据库(即将上线,数据库收录了美国FDA 过去十年近50000条检查信息和6000多封483报告原文,还可提供检查员信息等迎检定制服务),官方行动指示(OAI)最终转化为警告信的比例近似为20%(考虑到检查结果的升级和降级,这个比例是假设FDA合规办公室升级、降级策略基本不变的近似结果,但其实合规办公室近年策略有所变化,对结果的准确性有一定影响)。也就是说,当检查结果很不理想的时候,仍可能有80%的机会避免最坏的结果。
另一方面,2017年和2018年,尽管FDA生物制品审评与研究中心(CBER)检查结果为OAI后,发出警告信的比例相对稳定,药品审评与研究中心(CDER)却显得更为激进,2017年有一半不良的检查结果转化为警告信,而2018年这一比例达到了惊人的70%,也就是说,企业通过良好的483回复避免警告信的难度大大增加了。回复的专业性和整改措施的有效性,价值进一步增加了,那么识林从基础课开始,和大家一起从教训开始学习,看看常见的OAI缺陷,哪些回复被FDA明确认为是不好的。
常见缺陷:
本文仅以211.192调查不充分为例,列举被FDA认为效果不佳的回复。其它常见缺陷容易出现的不佳回复是怎样的呢?FDA期望的回复又是怎样的呢?更多分析请关注后续系列文章。也请关注IPEM课程。
不反省体系缺陷,强调个人问题:公司的锅,员工来背
[企业观点] 企业表示,该偏差是由于员工认为只要所有关键质量属性都符合质量标准,就可以继续压片操作并忽略异常设备控制值(例如,压片力,填充深度,AWC)。
[FDA观点] 企业的回复承认原来的调查是有限且不全面的,因此没有充分确定数据可靠性问题的范围和原因。企业的回复不充分,因为没有提供工艺参数(例如填充深度和压片力)高变异性的根本原因,以及为什么这些值与申报批压片过程中已证明的更为严格的控制不一致。企业没有充分说明异常值对批次质量的影响。
[企业观点] 企业表示创建这些记录的员工不再在该公司工作,并且这些原辅料在复检时合格。企业还报告说已更新了“良好文件记录规范”规程以及关于取样和放行原辅料的规程。
[FDA观点] 企业的回复不充分,因为没有提供容器和瓶盖的复检结果(包括原始数据和工作表)以证明其符合可接受标准。
[企业观点] 企业将此不合格归因于“未知的实验室错误”。企业声称这个含量偏低的检测结果是离群值,最可能的根本原因是分析错误。
[FDA观点] 对于产品本身的变异性正被评估的情况,如XX,离群检验是不适用的。对于忽略低含量结果或未知的实验室根本原因这一非特异性结论,企业没有给出足够的理由。
不反省体系缺陷,强调产品合格:哪错查哪,查完都合格
[企业观点] 企业承认原始样品应作为调查的一部分进行检验以确定根本原因并且承认最终根本原因不具结论性。企业分析了使用该API生产的产品批次的留样,发现这些样品符合质量标准。企业还提供了一份对2017年1月至2019年3月期间所生产产品的所有“被无效”OOS结果的回顾性审查。
[FDA观点] 企业的回复不充分。企业未附上支持性数据来说明对使用OOS调查所涉API批次生产的产品批次的留样检验。此外,企业还发现了另外9个OOS调查,其中根本原因判定和调查没有科学论证。企业没有扩大调查范围,以涵盖可能使用OOS物料生产的相关批次的留样。
[企业观点] 企业表示,只是在得到XX批准将微粒“再混合”回溶液后才恢复生产的。企业还表示,“可以肯定地说这是沉淀的粉末微粒而不是微生物污染”,因为检验表明没有微生物污染。
[FDA观点] 企业的回复不充分,因为未能提供对可见微粒污染的调查,并且企业依据的是这样的假设-有限的检验可以保证批次的其余部分是安全可供使用的。由于微生物污染不是均匀分布的并且在检测中难以发现,采用严格的上游控制来保证批次质量是至关重要的。
[企业观点] 企业表示该产品所有者对该缺陷产品进行了一项毒理学研究,结果发现该产品不存在风险。
[FDA观点] 企业的回复不充分。企业没有对使用这种反应性容器密闭系统的所有产品批次的稳定性进行评估,以确保其在货架期内符合所有质量标准,包括但不限于含量和杂质谱。
不反省体系缺陷,辩称缺陷无所谓:不会再犯或者卖美国的都合规
[企业观点] 企业表示现在未使用回收溶剂来生产XX。
[FDA观点] 企业的回复不充分。虽然企业在FDA检查开始时已不再使用XX作为XX生产用回收溶剂XX的供应商,但企业没有确定纠正措施来确保对执行可能影响药品质量的职能的所有合同商进行充分的质量监督。
[企业观点] 企业的回复表示,XX API批次XX和XX已被退货、返工并放行给非美国市场的客户。
企业的回复还表示,在2017年8月,企业采用了一种新的XX分析方法,该方法使用XX LC-MS/MS方法来取代容易出现错误OOS结果的XX LC-MS方法。企业未能对最初使用XX LC-MS方法(企业指出该方法次于新方法)放行的所有XX API批次(包括XX批XX)XX结果的可靠性进行核实。
[FDA观点] 在对此函的回复中,请提供:
- 一份针对效期内所有XX API批次的风险评估。
- 一份修正后的投诉处理规程,以及为确保所有投诉经过充分记录和彻底调查而已采取的任何进一步控制措施的细节。
- 关于被退货药品的接收和返工的规程。
- 所有使用新XX LC-MS/MS检测方法放行至美国市场的XX API批次的XX检测结果。
[企业观点] 企业打算替换受影响的XX仪器。
[FDA观点] 仅仅采购新仪器、安装新升级后的数据采集软件以及启用软件的各种功能是不够的。只有在实施了适当的程序和系统时,这些措施才有效,以确保按要求保存数据,以便质量部门能审核生产和控制数据以及相关的审计追踪,作为评估API是否符合所有既定中控标准和稳定性检验标准的一部分。
拍胸脯保证,拿不出实证:态度诚恳,数据贫乏
[企业观点] 对今后客户投诉的调查程序将包括质量管理部门所有留样的检查和文件记录。
[FDA观点] 企业的回复不充分。企业的回复未能对之前的投诉和相关调查进行回顾性审查以确定科学合理的根本原因。企业的回复也未包括任何纠正措施以防止再次发生,未评估是否有相关批次受影响。企业也没有表示将评估投诉处理程序。
[企业观点] 企业表示已经调查了容器缺陷,包括可能受此问题影响的其他药品。
[FDA观点] 企业未能提供支持性文件的副本。例如,企业没有提供一份XX批次的2-dram瓶装XX的批记录副本,企业声称其中包含完整的调查。
[企业观点] 企业的回复承认该片剂缺陷可能是由于多个根本原因,将继续调查此问题。
[FDA观点] 回复缺少关于顶裂片剂调查情况的更新细节。企业也未附上与调查相关的CAPA。
强调难做或已经做的不错:我已经很努力,请不看结果,看过程
[企业观点] 企业的回复表示NDMA很难被检出。
[FDA观点] 但是,如果企业已进行深入调查,可能会在残留溶剂色谱图中就发现提示NDMA存在的指标。例如,企业告诉FDA调查人员,企业知道在缬沙坦API残留溶剂色谱图中XX峰后疑似NDMA被洗脱的出峰位置处存在一个峰。在检测时,企业认为这个未鉴别的峰是噪声,而未进行进一步调查。此外,使用XX工艺生产的缬沙坦API验证批的残留溶剂色谱图,以及2012年(XX和XX)的XX显示,在XX峰后疑似NDMA被洗脱的出峰位置处至少存在一个未鉴别的峰。
[企业观点] 企业的回复还表示其不是唯一一家在缬沙坦API中发现NDMA的公司。
[FDA观点] 对于企业的情况,FDA样品分析发现,企业所生产的缬沙坦API中NDMA含量要显著高于其它企业生产的缬沙坦API中NDMA含量。由于有数据表明多种工艺生产的API中存在杂质,以及企业调查存在重大不足,FDA非常担心企业工厂生产的所有中间体和API中可能存在致突变杂质。
[企业观点] 作为对Quillivant XR溶出问题回复的一部分,企业对溶出试验方法进行了多次修改,与2013年FDA批准企业的新药申请(NDA)202100中被认为可接受的方法不同了。当企业用修订后的分析方法复检不合格批次时,一个批次仍然不符合质量标准。此外,企业的调查INV-16-222发现2014年之后生产的批次表现出比之前批次XX的趋势。
企业的回复表示,粉末样品处理过程中的XX是导致溶出方法变异性的一个因素。企业表示未在检验方法中充分指定或在方法验证中说明样品的制备。企业表示,当采用NDA 202100中的溶出检验方法时,复溶样品到开始溶出检验之间允许间隔至少XX,而不是迅速检验样品。
[FDA观点] 企业的回复不充分,因为未能对生产过程中可能导致产品质量不一致(例如,溶出性能)的变异进行迅速彻底的调查。企业还未能充分解决Quillivant XR暂未放行或已放行分销往美国的所有效期内批次的质量问题。
必读岗位及工作建议: - QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。
- QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。
- 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。
- 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。
适用范围:
本文适用于化学药领域的原料药生产,包括创新药和仿制药,适用于大型药企、跨国药企以及CRO和CDMO等企业类别,发布机构为国际通用标准。 文件要点总结:
原料药的生产质量管理规范强调了从质量管理到生产控制的全过程管理。首先,文件明确了质量管理的原则和机构职责,特别强调了质量保证和质量控制的重要性,并规定了自检、产品质量回顾以及质量风险管理的具体要求。在人员方面,规定了资质、培训和卫生要求,确保员工符合岗位需求。厂房与设施章节详细规定了设计建造、公用设施和特殊隔离要求,以保证生产环境的适宜性。设备章节则涉及设计建造、维护保养、校准和计算机化系统的要求,确保设备运行的可靠性。文件还特别提到了无菌原料药的生产特点,包括生产工艺、厂房设施设备设计、生产过程管理以及环境控制等,这些都是确保原料药质量的关键环节。 以上仅为部分要点,请阅读原文,深入理解监管要求。 |