|
首页
>
资讯
>
推动上市后变更全球互信,罗氏与 WHO、EMA 合作试点
出自识林
推动上市后变更全球互信,罗氏与 WHO、EMA 合作试点
2024-08-13
长期以来,全球制药行业面临上市后变更(PAC)审批的严峻挑战。根据一项涵盖18家全球大型制药企业、3年间145000多个PAC案例的研究,全球PAC审批流程普遍缓慢。尽管72%的PAC能在6个月内获得某些国家监管机构的批准,但这并不代表全球范围内的批准。这种不一致性迫使制药公司管理多个产品版本,对供应链管理构成巨大挑战。
在此背景下,近日一项由罗氏(Roche)领导的创新试点项目,得到WHO与EMA的联合支持,与48个国家监管机构合作,展示了全球统一PAC评估和审批机制的潜力。如果这个潜力兑现为全球协调机制,预期将显著提升PAC审批效率,为药企减压,保障药品供应连续性。
罗氏的试点:以EMA审评结论为基础,推动全球“互信”(Reliance)
在WHO和EMA的联合支持与指导下,罗氏启动了备受瞩目的药品上市后变更(PAC)信赖试点项目,其目标包括:1、显著缩短审批时间,从通常的2.5年大幅减至6.5个月;2、通过提交与EMA一致的注册资料,减少各个国家特有的注册要求,促进全球PAC监管趋同;3、通过共享EMA的审评报告和发补问题及回复,来提高审批的透明度。参与的监管机构需同意遵守审批时间表,接受EMA作为参考监管机构并认可EMA的审评标准,并依靠企业的分析报告书免除样品检验。
在启动阶段,罗氏慎重选择了一个治疗严重危及生命疾病的单克隆抗体的重大原液工艺变更作为试点,该变更的实施对产品持续供应非常关键。罗氏组建了一个包含CMC国际运营部和法规政策部的跨职能团队,负责详细规划试点项目并确立内外部的参与计划。EMA作为参考监管机构,而WHO作为关键利益相关方,共同参与了该试点计划的启动。此外,罗氏公司对该重大变更制定了详细的全球注册计划,该计划评估了试点对内部资源和药品供应的潜在影响,并考虑了PAC提交、批准时间表、平行提交多个变更的(parallel submission)可能性、全球实施策略及变更实施的宽限期等。
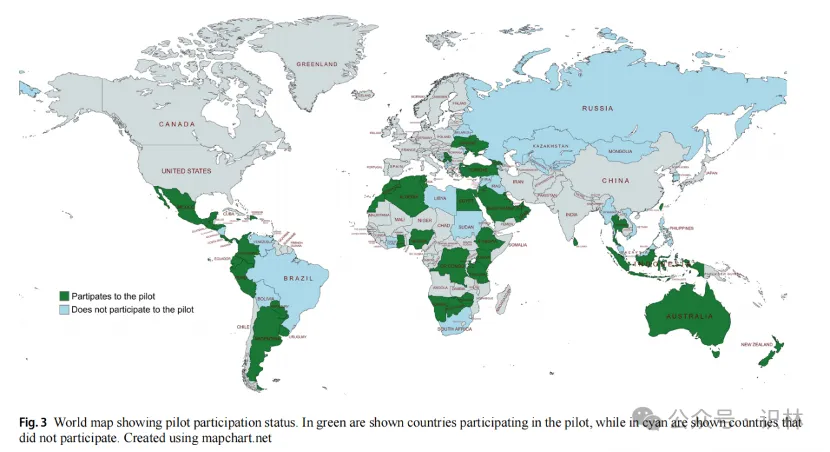
各国/地区药监的参与情况
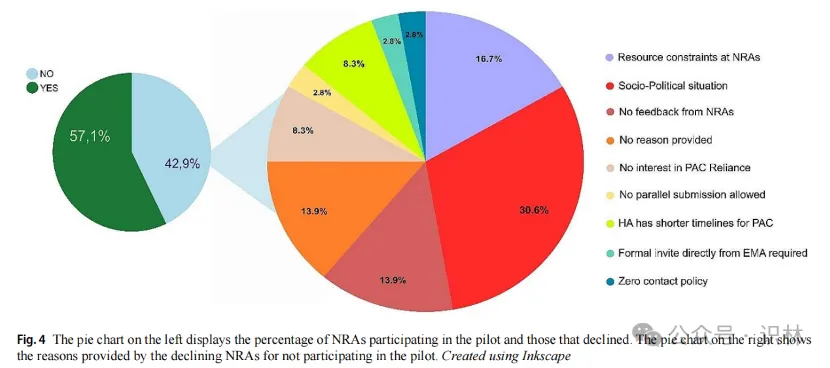
在罗氏接触的84个国家药监机构(NRA)中,有48家(57.1%)同意参与试点,31家拒绝,5家未明确答复(图1)。罗氏遵循了一定的选择标准,因此并未联系中国,美国,和日本,印度等监管机构。
NRA拒绝的原因多种多样(图4)。这包括:
- 8.3%因标准审批时间更短因此觉得没必要(蒙古和缅甸);
- 2.8%因现有的电子提交系统不允许平行提交多个变更(马来西亚);
- 30.6%因社会政治局势不稳定(巴勒斯坦、黎巴嫩、利比亚、叙利亚和委内瑞拉等);
- 8.3%因关注重点不同(韩国)8.3%;
- 13.9%未在规定时间内回应(阿鲁巴、巴西、毛里求斯),或
- 16.7%资源限制(阿尔巴尼亚、玻利维亚、北马其顿、伊拉克和南非)。
成果、经验与教训
克服了各种提交限制,罗氏在相应国家/地区的48家分支和代理商在一周内同步向NRA提交了申报资料。38家NRA有国家特有的注册要求,但在递交时,所有的NRA都接受了和EMA相同的注册申请资料,没有要求国家特有的注册资料。5家NRA最初可能将变更视为新注册,但通过企业的努力沟通,均同意与EMA保持一致,接受该申请作为上市后重大变更提交。罗氏还为满足多家NRA的要求,提供了西班牙语翻译。8家NRA原本不允许并行提交多个PAC,但其中6家还是同意了并行审评多个PAC。另有5家NRA通常不允许宽限期,但最终至少提供了6个月宽限期以保证产品的持续供应。
在这个颇为艰辛的过程中,罗氏获得了许多经验和教训,为将来可能的全球“互信”与协调,提供了宝贵的参考:
- (1) 透明度和对话:共享EMA批准的变更包和CHMP的审评报告是建立信任的关键。EMA的开放性促进了全球互信。
- (2) EMA和WHO支持:这两家机构的支持对试点至关重要,他们作为互信政策的倡导者,参与了启动会议并且EMA为参与该试点的NRA提供了解答审评报告疑问的机会。
- (3) 产品选择的重要性:选择对救命药供应有重大影响的变更,该变更的实施影响广泛的国际市场,是吸引NRA参与的重要因素。
- (4) 产品注册策略对供应链影响的评估:该策略必须考虑全球供应链、各国变更实施宽限期政策、是否允许并行提交、加速批准对供应计划的影响。各国不同的监管要求增加了评估和变更实施的复杂性,这进一步反映了全球监管体系协调的必要性。
- (5) 分支机构作用:当地分支与NRA的本地互动和沟通至关重要,尤其针对监管框架外的新工作方式,确保药企与NRA的信息一致性。
- (6) 监管环境多样性:84个国家和地区的不同监管环境造成诸多挑战,例如不同的变更分类、资料要求和批准时限。有的NRA觉得6.5个月已经比较长,而有的NRA通常需要几年才能批准上市后变更因此认为6.5个月太短。主要的挑战在于由于不同的变更分类和缺乏变更程序指南导致批准审评审批时限的不可预测性,这可能给全球药品供应带来挑战,进而影响患者继续获得药品。
应对全球PAC效率难题,仍然任重道远
变更难题普遍影响着制药行业,我国药监部门也积极采取措施应对。最近一个相当重要的政策变化,是业界非常关心的NMPA于2024年2月发布的《优化药品补充申请审评审批程序改革试点工作方案》,旨在提高审评审批质量与效率,通过整合省级监管资源,实现与国家监管的联动,为药品变更提供快速服务。试点方案聚焦化学药品,遵循"提前介入、一企一策、全程指导、研审联动"原则,确保审评审批的高标准和效率。方案还强调加强组织、审评、管理和技术支撑,以保障试点顺利和质量。
面对上市后变更审批的挑战,18家跨国药企的呼吁,罗氏的“互信”试点项目,WHO与EMA的支持,以及NMPA的补充申请改革试点均展示了行业与监管机构在提升药品PAC审评审批效能方面的积极探索与合作,从而逐步迈向国际合作、透明度提升、以及监管体系的优化,为药品的持续供应和患者福祉提供更好的保障。
参考文献
1. 8个方案应对上市后变更难题,18家药企巨头首席质量官联合呼吁
2. 国家药监局关于印发优化药品补充申请审评审批程序改革试点工作方案的通知
3. Unleashing the Power of Reliance for Post-Approval Changes: A Journey with 48 National Regulatory Authorities (https://doi.org/10.1007/s43441-024-00677-8)
特别感谢 IPEM 2014 级同学蔺亚萌在论文和本文中的贡献。
作者:识林-实木
识林®版权所有,未经许可不得转载
岗位必读建议: - QA:确保对药品补充申请审评审批程序的优化有充分理解,以支持药品生产技术迭代升级。
- 注册:熟悉改革试点工作方案,以便在药品注册过程中有效利用优化后的审评审批程序。
- 研发:了解试点工作条件,确保研发过程中的变更申报能够符合新的审评审批要求。
文件适用范围:
本文适用于化学药品的补充申请审评审批程序改革试点工作,由中国国家药监局发布,主要针对具备一定审评能力的省级药品监管部门。 文件要点总结: - 工作目标明确:旨在通过优化审评审批程序,提高药品监管和服务能力,缩短技术审评用时,提升审评审批质量和效率。
- 试点工作内容:以化学药品为重点,试点单位提供前置服务,包括指导、核查、检验和立卷服务。
- 试点条件严格:要求省级药品监管部门具备审评能力、管理制度、支撑保障等条件。
- 工作安排具体:包括试点准备、申报、确定试点单位以及开展试点工作的具体步骤。
- 保障措施到位:强调组织协调、公平公正、工作质量保障,确保试点工作顺利进行。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |