首页
>
资讯
>
阐明EC能否重构企业上市后变更之路
出自识林
2016-01-25 IPQ
2015年10月,ICH Q12 专家工作组(Expert Working Group,EWP)召开新一轮会议,探讨“已建立的条件”(Established Conditions,EC) 与 ICH Q12 在全球生命周期 管理中应起到的作用。对于“已建立的条件”这一术语,尚无权威的中译。US FDA 曾在2015年发布过一篇名为 Established Conditions: Reportable CMC Changes for Approved Drug and Biologic Products 的指南,文中称 EC 一词源自法规 21 CFR 314.70(a)(1)(i) 【见识林资讯 关于批准后变更的监管,FDA定义了“已建立的条件” 】而6月ICH专家工作组在日本召开的工作会,也是将FDA指南作为蓝本,对 EC 概念展开讨论。[1]
ICH希望企业与监管方沟通上市后变更可通过定义EC明确
生命周期 是较为宏观的概念,实际上涵盖了企业从产品申报 、建立工艺,到申请上市后变更 ,包括在质量管理体系 下实施持续改进等广泛的内容。EC 则可定义为“为保证获批产品的工艺性能和质量,而在申报资料中描述的产品、生产工艺、设施设备和相关控制策略(control strategy)集合。”在生命周期之中,显然会发生很多触及EC的变更,需上报药监审评。
药监制度较完备的国家,多有一系列法规、指南,针对不同程度变更,施加对应的审评或管理(部分示例如下表)。但长期以来,总有人认为“现行药品监管方法,对不同风险行为的区分不够有效,或阻碍了药业的创新”。而且在ICH框架下,即使近年来质量分类新发布的指南,Q8 - 11 ,强化了“质量源于设计”、“风险管理”等理念,强调了产品的特异性以及企业在生产药品过程中的知识积累;但讨论还是集中于药品开发的较早期阶段;对生命周期之中,获批上市以后产品的管理与工艺改进缺乏讨论。[2] 变更程度的界定在不同国家有别,具体的处置方式也不尽相同,比如我国有总局审批、省局批准以及省局备案等通道;US FDA 则划分为了 PAS 、CBE-30 、CBE-0 、Annual Report 等。这都体现了出台 Q12 与定义 EC 的必要性。
FDA先行试水,业界反馈不一
先行发布的FDA指南 Established Conditions: Reportable CMC Changes for Approved Drug and Biologic Products ,讨论的重点是:(1)申报资料中,哪些信息构成了“EC”(以普通的 CTD 格式为例探讨,上市后触及 EC 并超出了原变量范围的变更,必须报送FDA);(2)哪些 CMC 变更仅在企业质量体系下自行处置即可。该指南在60天内所收到的公开评议相当具体。这些问题多是讨论 ICH Q12 时也要面对的:
辉瑞(Pfizer)要求, FDA应阐明该指南与现行有关变更各指南的关系,如
国际制药工程协会(ISPE)指出,指南中将工艺验证排出所划定的 EC 范围,值得商榷
美国仿制药协会(GPhA)则表示,监管部门应给出更多具体的说明和例子,避免不同个人解释不同,尤其应避免审评人员之间尺度不同。此外,GPhA 还关注对审评资料要求更多的信息,是否会给新申请招致拒收 风险。
还有机构,如美国药品研究与制造商协会(PhMRA)指出, ICH在进行相近的工作,FDA应该加强与ICH工作组的合作,
参与编写该指南的FDA仿制药办公室官员表示,完善指南细节还有很多工作要做,但开始思考 EC 的定义,为ICH工作组作出了有益的铺垫。同时如业界所呼吁的,FDA也希望自己在这方面的努力与 ICH相一致。[3]
在FDA官员所参与的讨论中,有参会者提及FDA在管理变更的实践中,可比性协议 (Comparability Protocols)得到了良好的推广。FDA官员则介绍,可比性文件过去多用于生物制品,而且可能也带有过于宽泛、信息不足的缺点。但是FDA相关指南的更新已提上日程(详见FDA CDER Guidance Agenda 2016 ,FDA于上周末发布的2016指南修订工作计划)。或许随着此份指南的更新与 ICH Q12 的推进,可比性协议 能得以更广泛的应用。
虽然大多围绕 EC 的讨论均在强调产品的特异性,基于风险进行差异化的管理。但是也有参会者在强调药典——这一标准化工具的作用。比如来自德国的生物制品审评员 Martijn Van der Plas,他认为即使是最新、最复杂、有着诸多产品特异试验的新生物制品,依然会借助一般性的检验项目来控制;而这些项目中繁复的描述,包括接受限度 以及系统适用性试验 ,均已在药典进行了详细的介绍。所以药典在产品生命周期中仍扮演着重要的作用,至少可以作为CTD文件的引用源,节省申报资料的篇幅。
ICH Q12 草案或于2016年夏面世
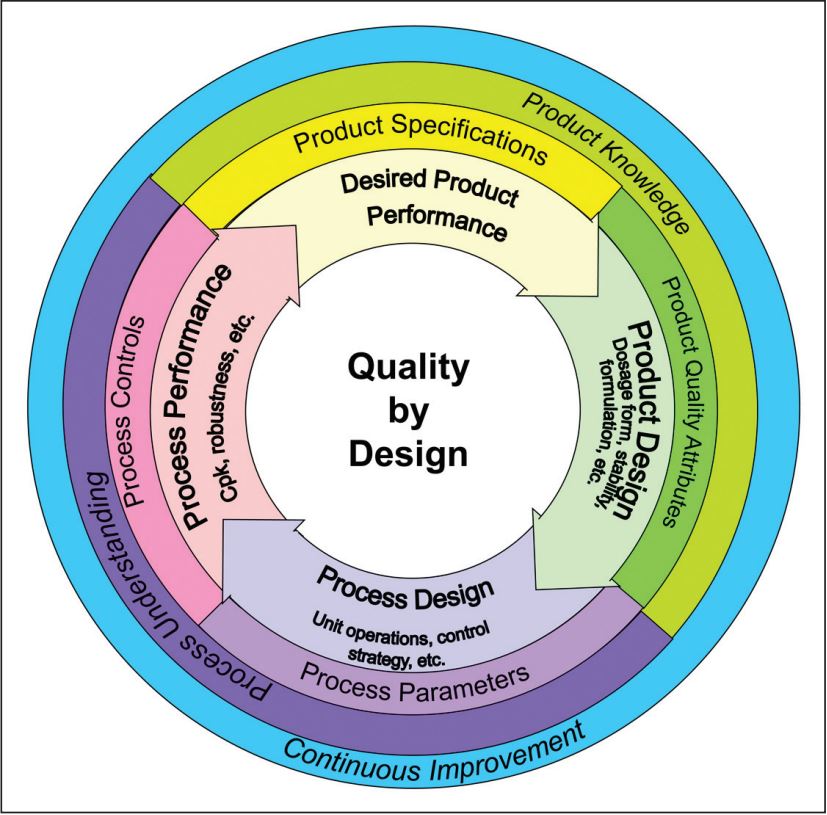
此次ICH协调委员会的会议书记是 Moheb Nasr,他是葛兰素史克的全球CMC战略副总。在加入葛兰素史克之前,他曾就职于FDA新药审评办公室(Office of New Drug Quality Assessment),参加了 ICH Q8 - Q11 系列指南的起草与实施,他对此类纲领性的文件驾驭能力不俗,比如这幅常被业界引用来阐释 FDA 有关QbD 理念的图示 (笔者将称其为”QbD 轮子“),正出自 Nasr 之手。按照ICH已公布的工作计划,Q12 的准备工作将于2016年夏走完第二阶段,届时将完成文件草稿并开始征求意见,而后呈递给欧美日各缔约监管部门签署实施。我们对此保持关注。
[FDA-2015-D-1659] 全文参考 1 EMA. How to improve product lifecycle management
[1] 2 IPQ. ICH Q12 EWG Views Established Conditions as Pivotal in Evolving Lifecycle Regulation Internationally; EMA Workshop Provides Input
[2] TM www.shilinx.com版权所有,未经许可不得转载。如需使用请联系admin@shilinx.com
法规指南解读:
适用岗位(必读):
工作建议:
QA:确保年度报告中记录的变更符合FDA指南要求。 注册:在提交年度报告时,注意包含所有适用的CMC变更。 生产:记录生产过程中的变更,并评估其对产品质量的潜在影响。 研发:在产品开发阶段考虑可能的变更,以减少后期的合规风险。 文件适用范围:
文件要点总结:
年度报告记录变更: FDA建议对于化学、制造和控制(CMC)的变更,如果预计对产品质量影响最小,应在年度报告中记录。
变更分类: 根据21 CFR 314.70,变更分为主要、中等和次要三类,不同类别的变更需采取不同的报告方式。
风险评估: 企业需基于风险评估方法,确定变更是否对产品质量有潜在不利影响,并据此决定变更的报告类别。
附录A和B的变更示例: 提供了在年度报告中记录的CMC变更示例,以及根据FDA的SUPAC指南和其他变更指南记录的变更示例。
CGMP合规: 无论变更如何报告,都必须符合CGMP(现行良好生产规范)的要求。
以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议 注册部门 :必须理解所有变更的法规要求,确保申报材料符合指南规定。质量保证部门(QA) :应熟悉变更对产品质量的潜在影响,并确保所有变更在实施前得到适当的评估和批准。研发部门 :在进行产品配方或生产工艺的变更时,需遵循本指南的推荐类别进行相应的申报。生产部门 :对于生产过程和场地的变更,应依据本指南进行操作并记录。文件适用范围 本文适用于化学药品的创新药和仿制药,包括原料药和生物类似药。适用于在美国进行注册的药品,由FDA发布,适用于Biotech、大型药企、跨国药企以及CRO和CDMO等企业类别。
文件要点总结 变更报告类别: 明确了变更的四种报告类别:重大变更需提交Prior Approval Supplement,中等变更分为两种情况,轻微变更则在年度报告中描述。 变更评估: 强调了对变更影响的评估要求,包括符合规格的一致性和可能需要的额外测试。 生产场地变更: 对生产场地变更进行了分类,并指出了哪些情况需要提交Prior Approval Supplement。 生产工艺变更: 详细列出了生产工艺变更的不同类别,并对需要提交的补充申请类型进行了规定。 规格变更: 对规格变更的管理进行了规定,明确了哪些变更需要通过Prior Approval Supplement提交。 容器封闭系统变更: 讨论了容器封闭系统变更的潜在影响,并规定了相应的报告类别。 标签变更: 对标签内容变更的管理要求进行了规定,包括需要提交Prior Approval Supplement的重大变更。 其他变更: 对不属于上述分类的其他变更进行了描述,并指出了相应的报告要求。 多重相关变更: 对于涉及多个变更的情况,建议根据最严格的报告类别进行提交。 以上仅为部分要点,请阅读原文,深入理解监管要求。
法规指南解读 适用岗位(必读) 研发(R&D) :负责药品配方和工艺的开发,需了解成分和组成变更的级别分类。生产(Production) :涉及药品生产规模变更,需遵循相应的测试和文件要求。质量保证(QA) :确保所有变更符合监管要求,负责审核相关测试和文件。注册(Regulatory Affairs) :负责提交变更文件和维护药品注册状态。工作建议 研发 :在进行成分或组成变更时,应根据变更级别选择合适的测试和文件。生产 :在规模变更时,确保遵循CGMP并进行适当的验证。QA :审核所有变更文件,确保符合监管要求,并监督测试的执行。注册 :了解变更的监管路径,准备并提交相应的补充申请或年度报告。适用范围 本文适用于美国FDA监管下的化学药品的立即释放固体口服剂型。涉及新药申请(NDA)、简化新药申请(ANDA)和简化抗生素申请(AADA)的变更,包括成分或组成、生产地点、生产规模变更以及生产过程和设备变更。
文件要点 变更级别定义 :根据对药品质量和性能影响的可能性,将变更分为三个级别,并为每个级别提供测试和文件要求。化学、制造和控制测试 :明确了每个变更级别的推荐测试,包括稳定性测试和溶出度测试。生物等效性研究 :对于可能显著影响药品质量和性能的变更(Level 3),要求进行全面的生物等效性研究。文件提交要求 :根据变更级别,规定了年度报告、生效中变更补充申请和预先批准补充申请的提交要求。监管路径 :提供了基于21 CFR 314.70(a)的变更通知的简化途径,允许在提交补充申请时或在下一个年度报告中通知变更。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
QA(质量保证):负责确保所有变更符合监管要求,并监督测试文件和记录的更新。 生产(Production):需要根据文件指导调整生产过程,并确保所有变更得到适当的验证。 注册(Registration):负责提交变更相关的补充文件和年度报告,确保所有文件符合监管机构的要求。 研发(R&D):在变更涉及新配方或新工艺时,负责开发和验证新的制造过程。 工作建议:
QA:监控变更实施过程,确保所有测试和文件符合监管要求,并及时更新SOPs。 生产:在变更实施前进行彻底的风险评估,并在变更后进行必要的工艺验证。 注册:准备和提交所有必要的补充文件,包括稳定性数据和溶解测试结果,并确保及时提交年度报告。 研发:在变更涉及新配方或新工艺时,进行必要的研发工作以确保产品质量不受影响。 适用范围:
要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
法规指南解读:ICH Q8 Pharmaceutical Development 适用岗位(必读) 研发(R&D) :深入理解药品开发过程中的科学方法和质量风险管理。质量管理(QA) :确保药品开发符合ICH Q8指南要求,建立设计空间和控制策略。注册(Regulatory Affairs) :在药品注册文件中准确呈现药品开发信息,包括设计空间和控制策略。工作建议 研发(R&D) :应用科学方法和质量风险管理来设计药品及其制造过程,确保产品质量。质量管理(QA) :监督药品开发过程中的质量控制,确保设计空间和控制策略得到有效实施。注册(Regulatory Affairs) :在CTD格式的注册文件中,合理布局药品开发相关信息,确保监管机构能够清晰理解。适用范围 本文适用于化学药品、生物制品、原料药等多种药品类型,包括创新药、仿制药、生物类似药等注册分类。适用于跨国药企、大型药企、Biotech等不同企业类别。发布机构包括中国、美国、欧盟等ICH成员国。
要点总结 设计空间(Design Space) :提出了设计空间的概念,强调了在设计空间内操作不视为变更,超出设计空间则需要启动监管变更程序。质量风险管理(Quality Risk Management) :强调了质量风险管理在药品开发过程中的重要性,特别是在识别和控制对产品质量有影响的关键因素。关键质量属性(Critical Quality Attributes, CQAs) :明确了CQAs的识别和控制是确保产品质量的关键步骤。控制策略(Control Strategy) :提出了基于对产品和过程深入理解的控制策略,包括对关键过程参数和物料属性的控制。生命周期管理(Lifecycle Management) :强调了在整个产品生命周期中持续改进和创新的重要性。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA:确保生产过程变更符合GMP规范和相关法规要求。 生产:在变更过程中严格遵循技术评价和验证流程。 注册:负责向SFDA提交变更申请及相关方案资料。 研发:提供变更后产品有效性、安全性的技术支持和数据。 文件适用范围:
要点总结:
变更原则 :所有生产过程变更应以提高产品安全性和有效性为出发点。变更申请 :生产企业需向SFDA提出变更申请,并提供证明资料。变更分类 :变更分为I、II、III三类,根据对产品质量影响程度进行分类。技术评价与验证 :所有变更均需进行技术评价,并根据变更类型进行相应的验证。变更内容与要求 :详细列出了主要原辅材料、菌毒种库、生产工艺、配制、成品、主要生产设备等方面的变更内容及其技术评价和验证要求。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA:确保药品补充申请注册事项符合法规要求,监督申报资料的完整性和准确性。 注册:负责药品补充申请的注册流程,确保所有申报资料符合要求。 研发:在药品规格、生产工艺等变更时,提供必要的药学研究资料和药理毒理研究资料。 临床:在需要进行临床试验的注册事项中,负责临床试验的设计、执行和资料提交。 文件适用范围:
文件要点总结:
补充申请注册事项 :明确列出了国家局审批、省级局批准及备案的补充申请事项,涵盖药品批准文号申请、商品名称使用、适应症增加、用法用量变更等。申报资料要求 :强调了申报资料的完整性,包括批准证明文件、证明性文件、修订的说明书和标签样稿、药学研究资料等。药学、药理毒理及临床试验资料 :对变更药品规格、生产工艺、组合包装等注册事项,提出了具体的研究资料要求。变更管理 :对于药品生产企业名称变更、生产场地变更、原料药产地变更等,规定了相应的申报资料和流程。说明书和标签修改 :对根据国家药品标准或监管要求进行的说明书和标签修改,提出了具体要求和流程。以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA:负责确保质量管理体系的实施和监督,建议定期审查和更新质量管理体系文件。 生产:确保生产过程符合质量管理体系要求,建议参与设备和工艺管理的持续改进。 研发:在产品设计和开发阶段考虑质量管理体系要求,建议与QA紧密合作以确保合规性。 适用范围:
文件要点总结:
质量管理体系概述 :明确了质量管理体系的发展、基本概念及其相互关系,强调了高层管理者在质量方针、目标和计划制定中的关键作用。产品质量实现要素 :涵盖了机构与人员、厂房设施、设备、物料与产品、工艺管理等关键要素,特别指出了人员培训和设备生命周期管理的重要性。质量保证要素 :包括变更管理、偏差管理、产品质量回顾、投诉和召回管理,强调了CAPA系统在持续改进中的作用。质量风险管理 :介绍了质量风险管理的职责、模式图、流程和步骤,以及在企业和管理机构中的应用。质量管理系统文件 :规定了文件体系结构、生命周期和种类,强调了文件管理在确保质量管理体系有效运行中的重要性。以上仅为部分要点,请阅读原文,深入理解监管要求。