首页
>
资讯
>
2019 年中国企业不合格 483(OAI)细读
出自识林
2020-01-14
注:在研究过程中,我们发现FDA提供的清单存在显著缺失,不少识林483数据库 中已经收录的483并没有在其中体现,FDA对此备注说:并非所有的检查都收录在数据库里,不包括州检查、批准前检查 、专论设施检查、等待最终执法行动的检查和非临床实验室检查。如果大家还发现有其它类型的缺失信息,欢迎向我们反馈,便于我们统一和FDA交流。
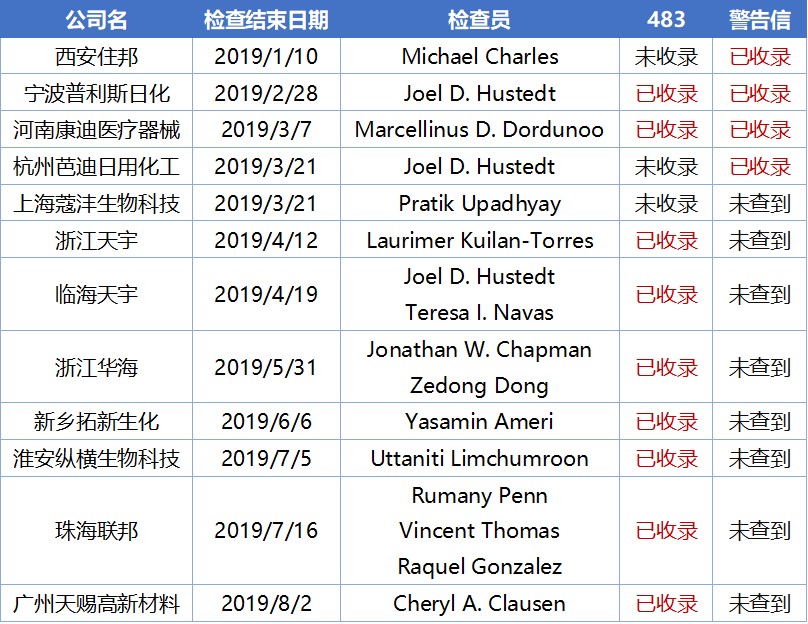
FDA公布的检查清单(https://www.fda.gov/inspection-classification-database ),收录了2019年的1027次药品GMP检查,其中表示检查未通过的OAI有62次,占6%;代表零483通过的NAI有387次,占38%;有缺陷通过检查的VAI有578次,占56%。其中中国企业接受检查99次,占不到10%,但中国企业检查不合格(OAI)有12次,占所有不合格的检查数的19%,也就是中国企业检查不通过的比例几乎是平均水平的二倍。
识林483数据库,收录了中国企业2019年这12次OAI中的9份483,下面就和大家一起细读一下这些2019年的经验教训。
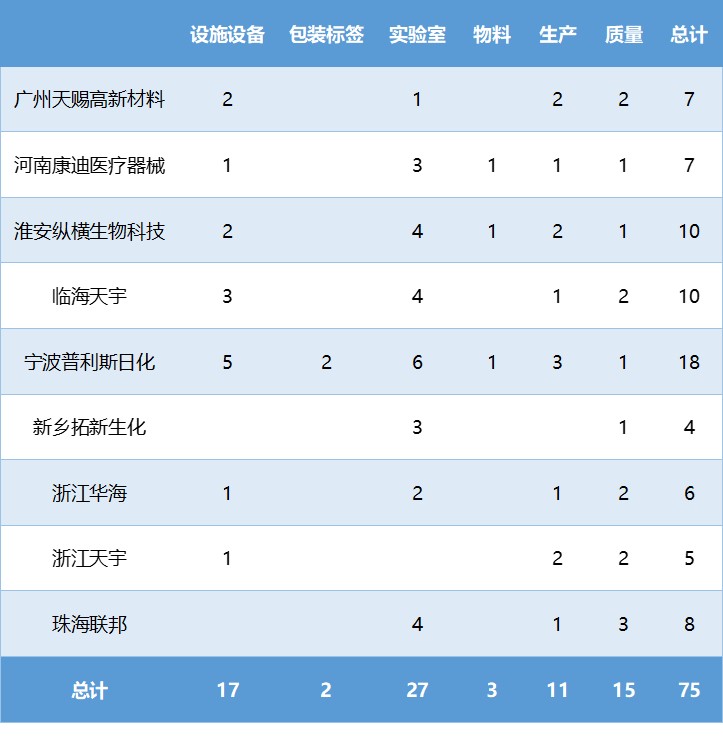
这9份483一共涉及75条缺陷 ,很难判断哪条缺陷是直接或最终导致OAI检查结果的原因,但通过系统的梳理和统计分析,仍然能找到不少值得思考的问题。
实验室仍是重灾区,杂质分析关注度不亚于数据可靠性
在所有75条缺陷中,实验室占27条,约36%,几乎覆盖了全部9份OAI。与预期不同的是,数据可靠性 相关的缺陷只有4条,最多的缺陷是实验室没有保证产品和其它成分符合既定的质量标准 ,有7条相关缺陷,但主要是提给OTC 类产品。
对原料药 企业,受亚硝胺 杂质问题的影响,分析方法验证 、杂质 、溶剂回收共有8条相关缺陷,比如关于气相色谱-质谱(GC-MS),FDA从杂质分析方法的特异性、干扰峰的处理、系统适用性方法的可靠性、样品溶液稳定性、GC-MS未知峰接受标准的科学性全面挑战了分析方法。
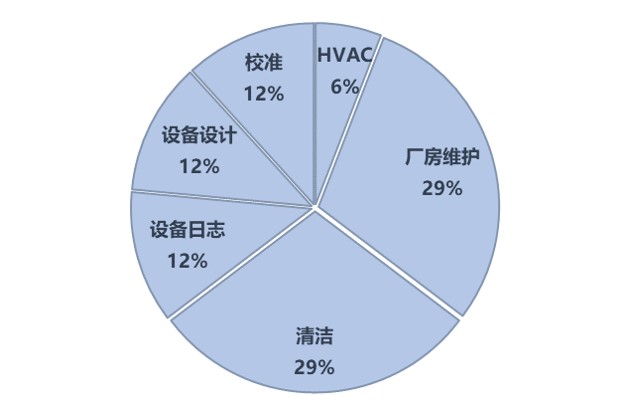
设施设备高居第二,FDA更关注“硬件”的“软肋”
关于设施设备的17条缺陷,仅有18%的设施设备缺陷关于硬件硬的地方(设备设计、HVAC),其余82%的缺陷是关于设施设备的运行和维护,尤其是清洁 和维护 ,合计占比接近60%。
设施 设备 缺陷主要集中在宁波普利斯日化等做OTC的“非正统”制药企业,FDA药品评价与研究中心合规办公室下属的生产质量办公室主任Francis Godwin(主管GMP合规)在2019年9月24日在IPEM师生讲座交流中特别强调,FDA在OTC方面近期格外关注,尤其向中国和韩国发出了大量OTC相关的警告信。这类企业,往往会在设施设备的“基础GMP”上出现问题,但华海、天宇等国际化多年的企业会出现什么样的问题呢?还是清洁:
A) 清洁方案的取样点没有合理评估,所有设备都是一个取样点;物料 和取样点的选择不是基于产品的理化特性、效价,毒性和清洁的难易程度,而是基于车间的生产计划;
清洁验证没有确定清洁后可连续生产的批次数;
阀门清洁的SOP 要求使用某种溶剂,但某次清洁记录没有相关溶剂的信息。
清洁和维护,相比之下可能是更为“高级”的缺陷,上面的五条具体案例,大家可以自查试试。
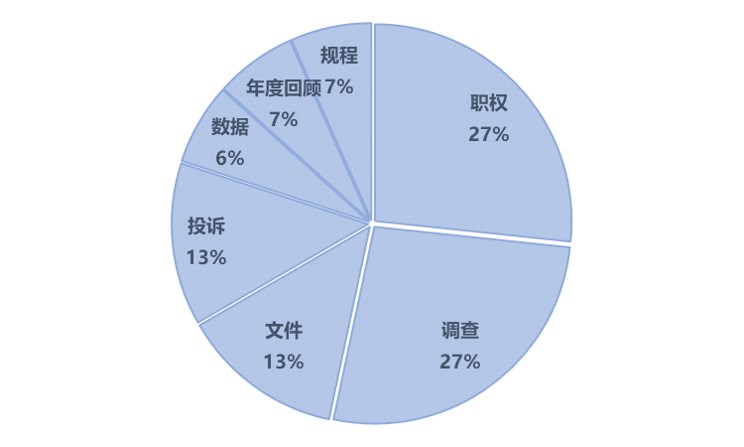
质量体系:调查和投诉考验企业体系成熟度
15条关于质量体系的缺陷中,质量部门的职权、调查、文件和投诉 是前四位的缺陷,占比超过80%。
很多缺陷都可以通过购买硬件设备、请咨询公司改善,但质量体系的有效运行,是一个需要从系统开始,不断累积数据、管理和培训的长期过程。见效慢、花费多,让企业更愿意把钱花到显示度高的地方,但“外功”补的快,钱到位就行;“内功”一旦拉下了,补起来就费时费力费钱。比如调查是FDA一贯重点关注的,但难以有效的减少。即使有十几年国际化基础的龙头企业,也不能简单避免这类问题,比如:用来监测步骤结束的薄层色谱(TLC)结果没有出来,就进入了下一步,后来Rf值明显有问题,没有做任何调查和CAPA(483的原文里好像有错别字,把preventive写成了presentative,细读者注) 。
另外,关于投诉的案例中我们可以看到:投诉是检查时的一条重要线索,相对温度和湿度在关于多晶型的投诉中被视为关键要素,但是CAPA中,并没有把针对多晶型的检验标准列入SOP,对X射线衍射方法也没做评估。也就是从投诉出发,检查员发现制定的CAPA并不能真正防止投诉的再次出现,反映出质量体系的不成熟。
工艺、验证和批记录是生产的关键
生产系统的缺陷中,有75%是关于工艺、验证和批记录。
比如关于溶剂回收的缺陷中,取样点的合理性没有经过论证且不能排除从后续工序中引入污染的可能性;另外一点值得参考的是,某种未知杂质,被证明在回收后的溶剂中高于回收前的,但这种异常的现象没有得到调查。
哪位检查员最爱发OAI?
2019年公布的中国企业OAI中,绝大部分检查员只参与了其中的某一次,但Joel D. Hustedt参与了宁波普利斯日化、杭州芭迪日用化工、临海天宇三次检查。他2007年加入FDA之前,曾担任新墨西哥州纳瓦霍地区印度健康服务和阿尔伯克基地区印度健康服务的环境健康专家;曾担任密尔沃基居民邮政的消费者安全官六年,负责药物和膳食补充剂 检查;担任明尼阿波利斯区麦迪逊居民邮政的州立联络员,现为FDA 驻华办公室药品检查员。
他是资深检查员,且较愿意发483,并有较高概率转化为警告信。可以查到的检查次数为163次,签发483(可查到)81次,签发警告信(可查到)11次,483发出率=483/检查次数=49.7%,警告信转化率=警告信/483=13.6%。关于他的背景、检查经历和历史检查记录整理,识林483数据库有详细的报告。
小结:OTC企业卡在入门问题,国际化资深企业体系欠成熟
限于篇幅,没有罗列更多缺陷细节,请登录识林483数据库查看更多,但没必要把数据库中的2000多份483都看了,到一定程度,FDA的检查逻辑就清晰了:找到体系中的异常情况(如偏差、OOS、未知峰、变更、投诉等),查看相关的控制策略 (CAPA),要求提供支持CAPA有效性的实证(数据),验证CAPA有效性的实效(是否重复发生),这四关走下来,体系就渐渐成熟了。
作者:识林-枫® 版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
必读岗位及工作建议:
QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。 QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。









{kind=link}