“请愿 ANDA”,在 1984 年 Hatch-Waxman 修正案颁布之前就已经存在,是一条通往 ANDA 批准的路径。在 Hatch-Waxman 法案颁布后的几年内,请愿ANDA 成为仿制药业药物研发范式的中流砥柱。尽管对于许多仿制药申请人而言,请愿 ANDA 仍然是不进行昂贵和耗时的临床研究而获得药品批准的可行路径,但近年来请愿 ANDA 受欢迎程度已经下降。请愿 ANDA 的持续成功和重新振作很大程度上取决于 FDA 能否在法定的九十天内迅速审查 ANDA 适用性请愿并采取行动。
本文将探索 ANDA 适用性请愿流程的好处,为什么不能产生预期作用,并对使程序回到正轨向 FDA 提出一些行动建议。
根据《联邦食品、药品和化妆品法案》(FD&C 法案)第 505(j)(2)(C) 节,ANDA 适用性请愿用于请求 FDA 许可提交与品牌参照上市药品(RLD)在规格、剂型、给药途径或复方药品中使用相同治疗类别的活性成分替代另一活性成分(当且仅当两种成分之间存在已知的等效剂量关系时可行)方面不同的 ANDA。多年来,FDA 已经收到了大约 1500 份 ANDA 适用性请愿。
FD&C 法案关于 ANDA 适用性请愿的条款很短,总共只有 150 多字:
如果某人想提交具有不同活性成分,或与参照药品在给药途径、剂型或规格方面有所不同的简化新药申请,此人应向卫生部长提交请愿,以寻求许可提交这样的申请。部长应根据本小节在请愿提交之日起九十天内批准或不批准请愿。除非发现以下情况,否则部长应批准请愿 —
(i) 必须开展研究以证明与参照药品不同的药品或其任何活性成分、给药途径、剂型或规格的安全性和有效性;或者
(ii) 根据所要求在简化申请中提交的信息,可能无法充分评估具有不同活性成分药品的安全性和有效性。
上述法规条款中加粗的一句话是我们关注的重点,因为 FDA 对适用性请愿的快速行动对于实现一个充满活力的适用性请愿计划至关重要。FDA 过去对适用性请愿的及时裁定记录一直不尽如人意。
这可能很大程度归结于2003年《儿科研究公平法案》(PREA),该法案修订了 FD&C 法案,增加第 505B 条,FDA 有权要求根据 FD&C 第 505 条提交新活性成分、新适应症、新剂型、新用药方案或新给药途径申请的申办人在儿科人群中开展测试。在活性成分、给药途径或剂型方面有所改变的适用性请愿 ANDA 会触发 PREA,因为 ANDA 也是一类根据 FD&C 法案第 505 条提交的申请。法规要求 FDA 拒绝适用性请愿,如果“必须开展研究以证明与参照药品不同的药品或其任何活性成分、给药途径、剂型或规格的安全性和有效性”。开展(甚至是推迟开展)儿科研究的要求触发了拒绝适用性请愿的法规要求。因此,除非 FDA 完全豁免 PREA 儿科研究要求,否则 FDA 必须拒绝请求许可提交与 RLD 在给药途径、剂型或活性成分方面有所改变的 ANDA 的适用性请愿。
对适用性请愿及其拟定改变的审查需要 FDA 药品审评与研究中心(CDER)多个部门的参与。虽然参与适用性请愿审查的 CDER 部门数量在一开始很少,但是参与部门数在过去几年中显著增加,导致这一程序难以控制。
在 Hatch-Waxman 修正案颁布之初,适用性请愿审查委员会只有来自仿制药办公室(OGD)的两名成员加上 Jim Bilstad 博士和 Bob Temple 博士(那时仅有的两名办公室主任)以及一名首席法律顾问办公室(OCC)的成员组成。OGD 起草了工作人员的工作建议并转发给负责 RLD 的相关 CDER 部门主管,并在月度适用性请愿委员会上做出决定。如果 CDER 部门主管没有反对,并且委员会的建议是批准请愿,那么最终决定权归委员会所有。如果 CDER 部门主管反对,或如果委员会的建议是拒绝请愿,那么将草拟拒绝函并由 OCC 代表审查,同时准备一封行动函。
而现在,插手的人实在太多了!有许多 CDER 办公室和部门主管需要在适用性请愿决定中表达意见。此外,除了极少数例外,有关适用性请愿先例的机构知识已经丢失。这进一步减慢了整个过程。然后是 FDA 的后 PDUFA 和后 GDUFA 环境,CDER 的大量注意力都集中在满足使用者费目标上。尽管 FD&C 法案要求 FDA 在提交后九十天内对适用性请愿做出裁决,但这并不是使用者费目标。因此,FDA 基本上是任适用性请愿自生自灭,而偏爱解决具有“更高”优先级的使用者费事项。
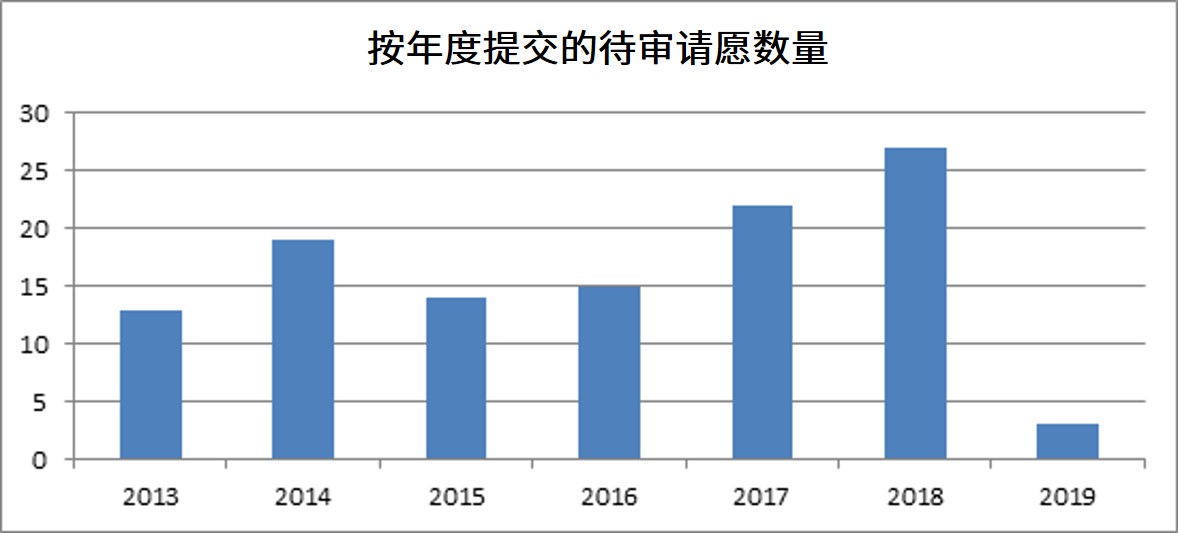
普享药协会(AAM,原仿制药协会)估计,目前有数百份 ANDA 适用性请愿正在 FDA 等待审理。其中一些请愿已经等待超过八年!根据 FDA Law Blog Citizen Petition Tracker 汇编的数据,FDA 在 2013 年至 2019 年间收到了 135 份 ANDA 适用性请愿。在这 135 份请愿中,有 113 份(84%)仍在等待审理(见下表)。在已经处理的 22 份请愿中,2 份(1.5%)被拒绝,5 份(4%)被许可,15 份(11%)被撤回(其中一半请愿后来重新提交,仍在等待审理)。
到现在为止,FDA 在解决 ANDA 适用性请愿积压问题上的努力尚未成功。FDA 于 2013 年 8 月发布政策与程序手册(MAPP)5240.5 确立了回复适用性请愿的政策和程序,并重申“FDA 将在请愿提交后不迟于九十天批准或拒绝请愿。”该 MAPP 并未导致 FDA 对适用性请愿的响应速度发生任何明显变化。FDA 于 2018 年 8 月修订了该 MAPP,删除了有关九十天期限的文字。自 2015 年 8 月以来,FDA 甚至没有更新 ANDA 适用性请愿跟踪报告。
也许是由于 FDA 未能及时处理适用性请愿,国会在 2017 年《FDA 重授权》(FDARA)法案第 805 条中表达了其对 FDA 达到九十天目标的期望。国会希望鼓励 FDA 加快对此类请愿的回复,要求 FDA 提供一份尚未解决的适用性请愿数量的报告和一份在提交 180 天后仍未解决的适用性请愿数量的报告。此外,GDUFA II 绩效目标函也指出“FDA 希望以更及时和可预测的方式回复适用性请愿。”但到目前为止,FDA 未遵守国会要求和 GDUFA II 绩效目标。
由于当前 ANDA 适用性请愿程序的失效,企业转而使用 505(b)(2) 路径,因为 505(b)(2) 涵盖了 ANDA 适用性请愿所允许的相同类型的改变。但提交一件 505(b)(2) NDA 不仅意味着支付高昂的申请使用者费,(在某些情况下)还意味着昂贵的年度项目费。
如何修复 ANDA 适用性请愿程序?
我们认为 FDA 有两条路可以解决适用性请愿积压问题,并为未来的成功奠定基础。
第一条路,FDA 可根据法定和监管要求以及 FDA 可用来作为最终决策过程基础的先例,将工作人员的工作外包出去制定建议。这种方法可能会让一些人感到不满,因为私人承包商可能会有与其潜在关系相关的利益冲突。
第二条路,或许是最好的方法,FDA 选取少数几个人花费大量时间管理适用性请愿程序,目的是清理积压并建立起未来可在 FDA 内部处理请愿的基础。让拥有历史视角的人来做这件事应该对 FDA 有最大的好处,他们的智慧也可以传递给 FDA 内的其他人。这个“适用性请愿突击小组”可能能够在为 FDA、患者和行业提供应有服务的同时,恢复请愿程序的活力,并为继续推进这一程序提供长期解决方案。
如果 FDA 没有能够在内部解决这一问题,国会可能不得不介入,并对法规做一些修改。例如,国会可能会考虑延长 90 天的响应截止期限,以反映 FDA 更加可能实现的最后期限,例如 150 天、180 天或国会近年来提出的 270 天请愿响应期限。
一个可实现的最后期限将为 ANDA 适用性请愿程序提供更大的确定性,打消仿制药业的顾虑,使他们了解到这是获得 ANDA 批准的可行且实用的路径,并且可能引发重新提交 ANDA 适用性请愿的兴趣。
作者:Kurt Karst,FDA Law Blog;Bob Pollock,Lachman Consultants
编译:识林-椒
识林®版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。