首页
>
资讯
>
EMA《药物警戒的影响评估策略》导读:不谈PV怎么做,而谈PV该怎么做才有效
出自识林
EMA《药物警戒的影响评估策略》导读:不谈PV怎么做,而谈PV该怎么做才有效
2022-05-06
2022年4月19日,欧洲药品管理局(EMA)药物警戒风险评估委员会(Pharmacovigilance Risk Assessment Committee,PRAC)发布了《药物警戒的影响评估策略》
这是一份药物警戒 监管领域的高阶文件,关注的是整个 PV 监管体系本身,而不是单个流程或单个药品。
药物警戒,并非只是到点提交安全报告,其最终目的是用药的安全和有效。目前,各国药物警戒的监管流程,以最简化的角度来看,通常是监管部门采集不良反应 信息(往往比较被动),分析信息,然后采取行动,如风险最小化措施(risk minimisation measures,RMM),可能包括改说明书,召回 等等,这样的做法是否真正有效?效果能否可量化?如果不够有效,那问题到底出在哪里?是采集的信息本身不完整?RMM 本身无效?还是实施者(如医生或药厂)执行不到位?接下来,又该怎么改进?
EMA 的这份文件,就在尝试回答上述一系列问题。对于我国 PV 监管者,企业 PV 部门的管理者,亦或是一线的 PV 人员,阅读本文,都可获得更多的视角,更广的视野。
以下是识林的简要导读,报告全文 可至识林查阅。
该文件描述了影响评估的概念方法、目标和原则,以及欧盟药品监管网络(EU Medicines Regulatory Network,EMRN)实施的影响研究(impact research)的优先顺序和流程,以系统地调查药物警戒活动和主要监管行动如何转化为可衡量的积极效果(例如,通过减少不良反应 造成的伤害),并确定可能抵消风险最小化措施(risk minimisation measures,RMM)的意外后果。
此次的第二次修订版整合了自发布以来五年间在四个领域取得的成果:
对PV概念的简要回顾
药物警戒 (Pharmacovigilance,PV)是发现、评估、理解和防范与药物有关的不良作用或其他任何可能与药物相关的安全性问题的科学活动。监管部门根据新出现的药物警戒数据采取的监管行动,旨在改变个人(即患者、消费者、护理人员和医疗保健专业人员)和医疗机构的认知和行为,有助于保护和促进公共健康,防止药物造成的伤害,确保药物能够安全有效地使用。
药物警戒活动由监管机构和制药公司执行,包括风险管理规划(risk management planning,RMP)、收集和管理可疑不良反应 (Adverse reaction,ADR)报告、信号检测和管理,以及上市后研究,等等这些活动,共同构建上市药物的安全用药信息。
PV体系有效性影响评估的概念方法、目标和原则
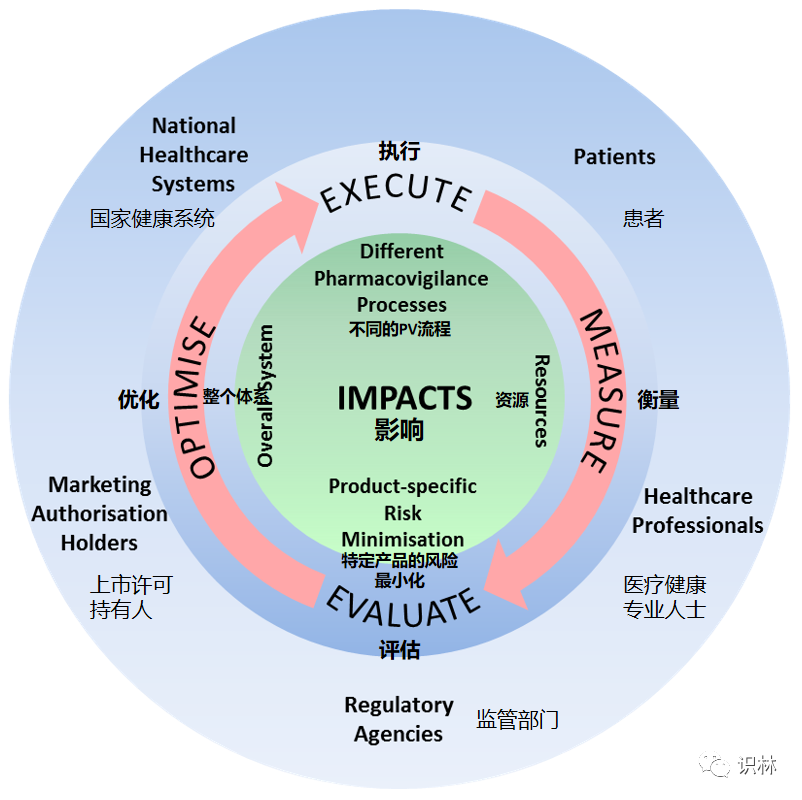
衡量药物警戒活动和监管行动影响的概念方法如下图所示,监测重要的药物警戒活动和监管行动,并衡量其对患者相关健康结果的影响,改善药物警戒系统 的功能。
药物警戒影响研究是监测药物的获益和风险并持续改进药物警戒系统功能的关键支柱。欧盟监管机构开展影响研究,生成针对特定产品或治疗类别监管行动的数据、信息和知识,并促进患者和医疗保健专业人员的参与,其目标是:
1. 通过评估风险最小化措施(RMM)的有效性,明确已采取重大风险最小化措施的授权药品(authorised medicinal products)的收益和风险;
2. 识别成功的药物警戒活动,以及还需加强的活动,并识别有效药物警戒系统 的推动因素和障碍。
为实现这些目标,该策略的概念方法以下列影响研究的指导原则为基础:
——以健康结果为导向;
——基于科学证据,采用稳健的方法;
——将核心药物警戒活动整合进欧盟药品监管网络和机构;
——与监管机构、学术界、患者和医疗保健专业协会合作开展;
——聚焦与患者获益高度相关的关键药物警戒活动;
——以风险相称的方式,专注于特定产品或治疗类别的监管行动。
该如何衡量PV活动的影响?四大关键活动领域
自2016年启动以来,PRAC 影响策略已通过四个关键活动领域(见下图)实施,最初的重点是制定方法指南和建立监管框架,以优先考虑和开展影响研究。2017年首次公开听证会启动了定性研究(qualitative research),更好地了解患者和医疗保健专业人员参与药物警戒活动的推动因素。2019年启动了对制药企业申办的上市后安全研究(Post-authorisation safety studies,PASS)的审查,评估风险最小化的有效性,探索药物警戒流程的改进方案。
PRAC 专题工作组 (Interest Group,IG)负责监督该策略的四个活动领域。
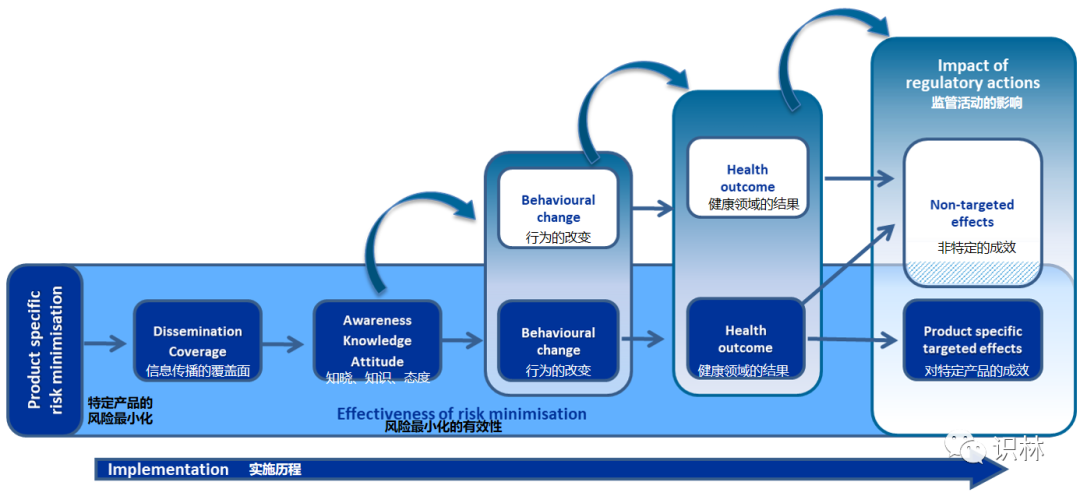
1. 风险最小化措施(RMM)的有效性评估
根据药物警戒质量管理规范(GVP)模块XVI 风险最小化措施:工具和有效性指标的选择 上市许可持有人 (MAHs)执行并向主管部门报告上市后安全研究(PASS),评估授权 RMM 的有效性。
该有效性评估基于对产品特定的目标成效的估计,以及潜在的非目标成效(例如,由于监管行动导致的处方、健康结果或医疗实践的意外变化),指导监管部门决策,即是否在风险管理规划(RMP)中加强或停止 RMM。例如,一项特定产品的 RMM 可能导致其他药品出现其他认知、行为或与健康相关的结果(例如溢出效应),从而抵消该 RMM 有效性。
系统的收集和回顾 MAH 申办的评估RMM有效性的 PASS 结果,有助于更好地理解:(1)数据收集、研究设计和分析方法 的要求,(2)对研究结果的解释,(3)与 RMM 成功或失败相关的因素和(4)RMM工具在临床实践中的影响。
具有重大公共卫生意义的监管行动的影响研究可以监测风险最小化的结果。
例如,2013年欧盟因心血管风险对含双氯芬酸的药品进行了标签变更,荷兰、英格兰和苏格兰的双氯芬酸使用量显著下降,但丹麦则没有,这与改用其他止痛药(如非甾体抗炎药(NSAIDs)、扑热息痛、阿片类药物等)有关。
此外,2015年因心血管风险对含羟嗪的药物进行了标签变更,英格兰和苏格兰羟嗪类药物的使用量立即下降,但对荷兰和丹麦的影响有限,仅对某些年龄组有影响。这一次,监管行动与改用其他抗组胺药、苯二氮卓类药物或抗抑郁药无关。
2. 特定药物警戒过程的有效性
药物警戒活动涉及多个复杂的过程,包括可疑不良反应 的自发报告、信号检测和管理、风险管理 规划、定期安全更新报告 和上市后研究。
2018年 PRAC 将衡量风险最小化有效性的上市后安全研究(PASS)确定为与该策略关键活动领域最相关的过程。自2012年以来,PRAC 已实施或要求进行大量的 PASS 评估 RMM 的有效性。然而,对 PRAC 在2016年至2019年期间评估的 PASS 进行系统评价,结果显示质量和方法存在明显的异质性 (heterogeneity),这可能是该系统性评价(systematic review)中近一半研究无法得出 RMM 是否有效的原因。对评估 RMM 有效性的 PASS 应进行持续审查,重点关注导致 PASS 没有结论或 RMM 无效的因素。
3. 有效药物警戒和利益相关方参与的推动因素
药物警戒流程和 RMM 的能否产生效果,最终还是取决于患者和医护人员,这些利益相关方在监管层面和日常医疗保健应成为药品监管和药物警戒活动的“推动者”。
他们在一些药物警戒活动中起着至关重要的作用,例如,通过报告可疑的不良反应 来提供信息,以及通过行为转变来实施 RMM。具体而言,在医药产品的风险管理规划和收益-风险生命周期 管理过程中,需要利益相关方为 RMM 的监管决策提供信息。
对2017年丙戊酸盐公开听证会上利益相关方的意见进行内容分析,从 RMM 实施的角度确定了这些意见的价值和不足之处。进一步的定性研究,是通过概念和风险治理框架分析了重大安全问题中的药物警戒活动,并为加强监管机构与患者和医疗保健专业人员之间的对话提供了建议。
4. 影响研究的分析方法
在衡量药物警戒活动的影响或评估风险最小化措施的有效性方面,没有一种普遍接受的方法。
2017年,一项对衡量监管干预措施影响的系统评价显示,研究的开展和报告存在明显的异质性 ,从而有必要为利益相关方提供关于影响研究方法的科学指导。欧洲药物流行病学和药物警戒网络中心(ENCePP)《药物流行病学探究方法学标准指导手册》第7次修订版包括一个专门用于影响研究的附录,该附录现已完全纳入当前版本。GVP 模块 XVI 包括 RMM 有效性评估的原则、目标,以及有效性评估方法的新附录,供制药行业和监管机构参考。随着影响研究领域的发展,影响研究分析方法 的指南也将不断更新。
影响研究的优先顺序该怎么定?
根据 GVP 模块 XVI 中 RMM 有效性评估的原则,监管影响研究(regulatory impact research)应侧重于重点患者和对公共卫生具有重要意义的监管行动,同时考虑风险的性质、严重性、人群暴露的程度和公众关注的程度。
PRAC 已经建立了一个影响研究的优先顺序和监管跟进流程,其中,优先顺序的评价标准中包含了监管行动的公共卫生重要性、对临床实践的潜在影响以及是否可以生成新的与决策相关的数据。
需要明确了解研究问题(即需要哪些数据或信息)、如何使用研究产生的数据(即研究是否有望减少不确定性)、研究的可行性和研究结果的普适性。
确定优先顺序使监管机构能够将资源用于需要额外证据的主要监管行动中,这些额外证据超出了常规药物警戒 流程(即,快速和定期的安全报告、上市后安全研究或包含在欧盟 RMP 中的衡量 RMM 有效性的研究)产生的数据。
外包给第三方开展 PV 监管影响研究
国家主管部门和机构有法律义务监督 RMM 的结果和授权药品的其他监管行动([DIR Art 107h (1) , REG Art 28a ]),虽然 MAH 产生的科学证据仍然是监管评估的核心,但可能需要额外的数据和信息,进一步为基于现有最佳科学证据的监管决策提供信息。
药物警戒影响研究是一项由多个利益相关方共同参与的工作,涵盖衡量欧盟药物警戒活动和监管行动对成员国国家医疗环境的影响。通常包括治疗类药物的多个上市许可,例如,在欧盟转介程序中采取的监管行动。在这种情况下,集中开展影响研究具有多方面的优势,例如,获取与影响相关的健康数据、说明各国在医疗实践中的差异、监管沟通策略和实施产品特定的监管行动等。
欧盟与第三方研究机构建立了框架合同,进行上市前和上市后的质量、疗效和安全性研究,生成数据和信息以支持监管决策。根据该框架合同,药物警戒活动和监管行动影响的定性研究和药物流行病学研究可能会委托给入围的外包机构(contractors)。PRAC 对委托的影响研究的研究问题、目标和方法学方面提供建议,并协助评估外包机构提交的研究方案、研究报告和出版手稿。
此外,2022年启动的数据分析和真实世界询问网络(Data Analysis and Real-World Interrogation Network , DARWIN)将利用来自欧洲各地的真实世界证据,对药物的使用、安全性和有效性进行影响研究,并扩大医疗数据库和标准分析目录。
欧盟成员国的主管部门可以自行开展影响研究或在 EMRN 内建立研究合作。例如,遵循多数据库研究的通用协议,每个监管机构在国家或地区数据库中进行研究,或通过将来自不同数据库的数据转换为通用数据。EMA 与西班牙和英国的国家机构合作进行了一项研究,评估了2013年欧盟标签变更 对儿童使用可待因治疗疼痛适应症 的影响,以及在2015年欧盟合作研究引入可待因 RMM 后儿童疼痛和咳嗽替代疗法的变化。
所有公立(publicly funded)的影响研究必须完全遵循 ENCePP 行为准则,以确保最大程度的科学独立性和透明度,并根据 ENCePP《药物流行病学探究方法学标准指导手册》的附件2遵守最高的方法学标准。
作者:识林-红木
识林® 版权所有,未经许可不得转载。