首页
>
资讯
>
FDA 向国外发出的警告信四分之三涉数据可靠性
出自识林
2019-08-13
昨天的资讯介绍了企业如何换个角度从监管视角审查自身问题,在监管检查之前发现隐藏的数据可靠性 缺陷。今天我们从一些警告信观察项数据以及资深专业人士的建议来继续看看如何及早、高效的发现数据可靠性问题。
前 FDA 检查员、Parexel 咨询公司副总裁 Ron Tetzlaff 于 2019 年 3 月在乔治亚大学第 43 届年度国际 GMP 会议 上分享了他在数据可靠性(DI)检查方面 40 多年的经验。Tetzlaff 在会上的演讲包括他对 FDA 药品审评与研究中心(CDER)生产质量办公室(OMQ)针对国外生产场地发布的警告信的深入分析。
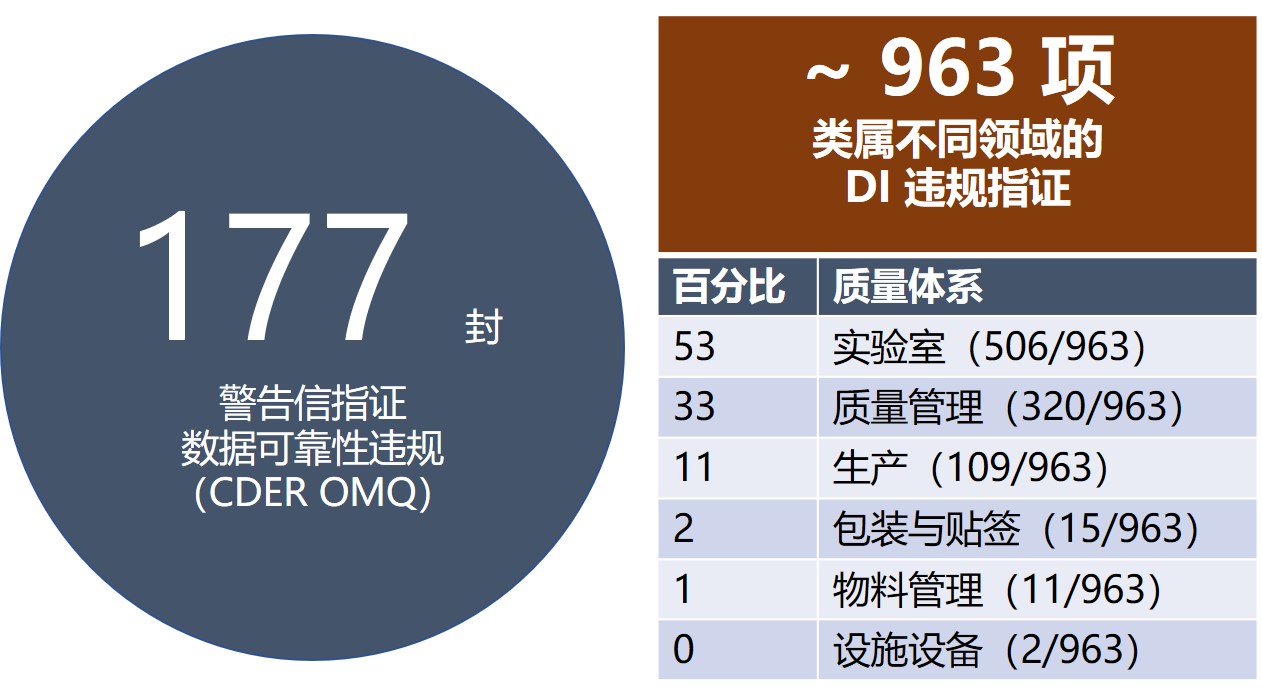
2012 年至 2018 年 OMQ 共向国外场地发布 239 封警告信,其中涉及数据可靠性的有 177 封,如下表。可以看到的趋势是,从 2012 年到 2018 年警告信数量显著增加(翻了一倍多),指证数据可靠性问题的警告信百分比一直保持在 75% 左右。
年份
总警告信数量
涉及 DI 的警告信数量
DI百分比(%)
2018
53
42
79
2017
61
47
77
2016
44
32
73
2015
20
17
85
2014
18
14
78
2013
21
10
48
2012
22
15
70
7年总计
239
177
74
在这 177 封涉及数据可靠性问题的警告信中,共有约 963 项指证,按系统划分, 53% 关于实验室,33% 关于质量管理体系 ,11% 关于生产,如下图。
他还分析了在这些警告信中,按系统划分的指证(citations)出现频率(见下表)。到目前为止,最常见的指证是 QA 监督。其次是没有原始数据 ,Tetzlaff 表示,如果 FDA 在警告信中明确指出原始数据被丢弃,则被归类为“原始数据-丢弃”,如果没有说明,则被归类为“原始数据-不可得”,这两类指证加起来大约相当于全部指证项的三分之一,都涉及到无法获得原始数据,无论是故意丢弃还是不知道什么原因的无法获取。
类别
全部质量体系
实验室
QA
生产
包装与贴签
物料管理
设施设备
审计追踪
53
53
/
/
/
/
/
文件管理
28
9
11
5
3
/
/
身份验证
9
/
/
1
2
4
2
QA 监督
271
/
271
/
/
/
/
原始数据-权限
72
66
2
4
/
/
/
原始数据-准确性
92
68
/
18
6
/
/
原始数据-备份
7
7
/
/
/
/
/
原始数据-丢弃
92
81
/
11
/
/
/
原始数据-伪造
71
51
/
19
1
/
/
原始数据-GDP
31
15
/
16
/
/
/
原始数据-不可得
142
101
/
33
1
7
/
拒绝提供数据/记录
36
/
36
/
/
/
/
SOP
17
13
/
2
2
/
/
试进样
42
42
/
/
/
/
/
总计
963
506
320
109
15
11
2
Tetzlaff 将拒绝提供数据/记录也归为数据可靠性问题的一个类别。他特别提到一封发送给日本企业的警告信 ,根据在识林警告信数据库检索,该公司应该是日本精化株式会社(Nippon Fine Chemical. Co)。这封警告信中提到,QC 管理层指示公司雇员肩并肩站在一起,阻止检查员进入实验室的部分区域以及接近分析销售至美国药品的设备。因此,Tetzlaff 认为拒绝检查是一个相当严重的问题。FDA 也将拒绝检查视为产品掺杂【阻碍GMP检查=产品掺杂 2014/10/24】 。
另外他还提到按国家和地区分类的数据可靠性相关警告信数量和分布,这在【2018 年指证数据可靠性问题的 FDA 警告信情况 2019/06/21】 一文中有更详细的统计,不再赘述。
在问答环节,Tetzlaff 被问及在时间有限时,可以采取哪些措施来发现 DI 问题。他表示,“能做的最好的事情就是走进现场,观察实际操作,看看操作人员正在做的些什么。”这一建议与识林昨天的资讯【企业应从监管视角审查自身数据可靠性 2019/08/12】 中 Mark Newton 的观点类似。Tetzlaff 表示,“我发现,大多数数据可靠性违规行为都不是在深奥的复杂领域,而是在人们日常手头工作中。”他也列举了一个同步记录的例子,“最常见的问题之一是他们不会在做的同时写下数据或结果。你看到他们在称重,但他们没有写下任何东西。没有电子记录,现场也没有任何记录,虽然他们称重了六个样品。没有人注意他们没有写下重量。然而,第二天你回去查看那条记录,记录就已经填写完成了。”所以,到现场去,将你从会议、报告或者警告信中学到的东西应用在现场观察上,你或许可以发现并改善大部分数据可靠性问题。Tetzlaff 表示,“事实上,我作为咨询顾问发现的所有问题都是显而易见的。”
作者:识林-椒® 版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
参考资料 识林资讯:2018 年指证数据可靠性问题的 FDA 警告信情况 2019/06/21 识林资讯:企业应从监管视角审查自身数据可靠性 2019/08/12 识林资讯:阻碍GMP检查=产品掺杂 2014/10/24
必读岗位及工作建议:
QA:负责确保质量管理体系的实施和监督,建议定期审查和更新质量管理体系文件。 生产:确保生产过程符合质量管理体系要求,建议参与设备和工艺管理的持续改进。 研发:在产品设计和开发阶段考虑质量管理体系要求,建议与QA紧密合作以确保合规性。 适用范围:
文件要点总结:
质量管理体系概述 :明确了质量管理体系的发展、基本概念及其相互关系,强调了高层管理者在质量方针、目标和计划制定中的关键作用。产品质量实现要素 :涵盖了机构与人员、厂房设施、设备、物料与产品、工艺管理等关键要素,特别指出了人员培训和设备生命周期管理的重要性。质量保证要素 :包括变更管理、偏差管理、产品质量回顾、投诉和召回管理,强调了CAPA系统在持续改进中的作用。质量风险管理 :介绍了质量风险管理的职责、模式图、流程和步骤,以及在企业和管理机构中的应用。质量管理系统文件 :规定了文件体系结构、生命周期和种类,强调了文件管理在确保质量管理体系有效运行中的重要性。以上仅为部分要点,请阅读原文,深入理解监管要求。