首页
>
资讯
>
KASA — 面向21世纪的药品评价模式
出自识林
2019-04-29
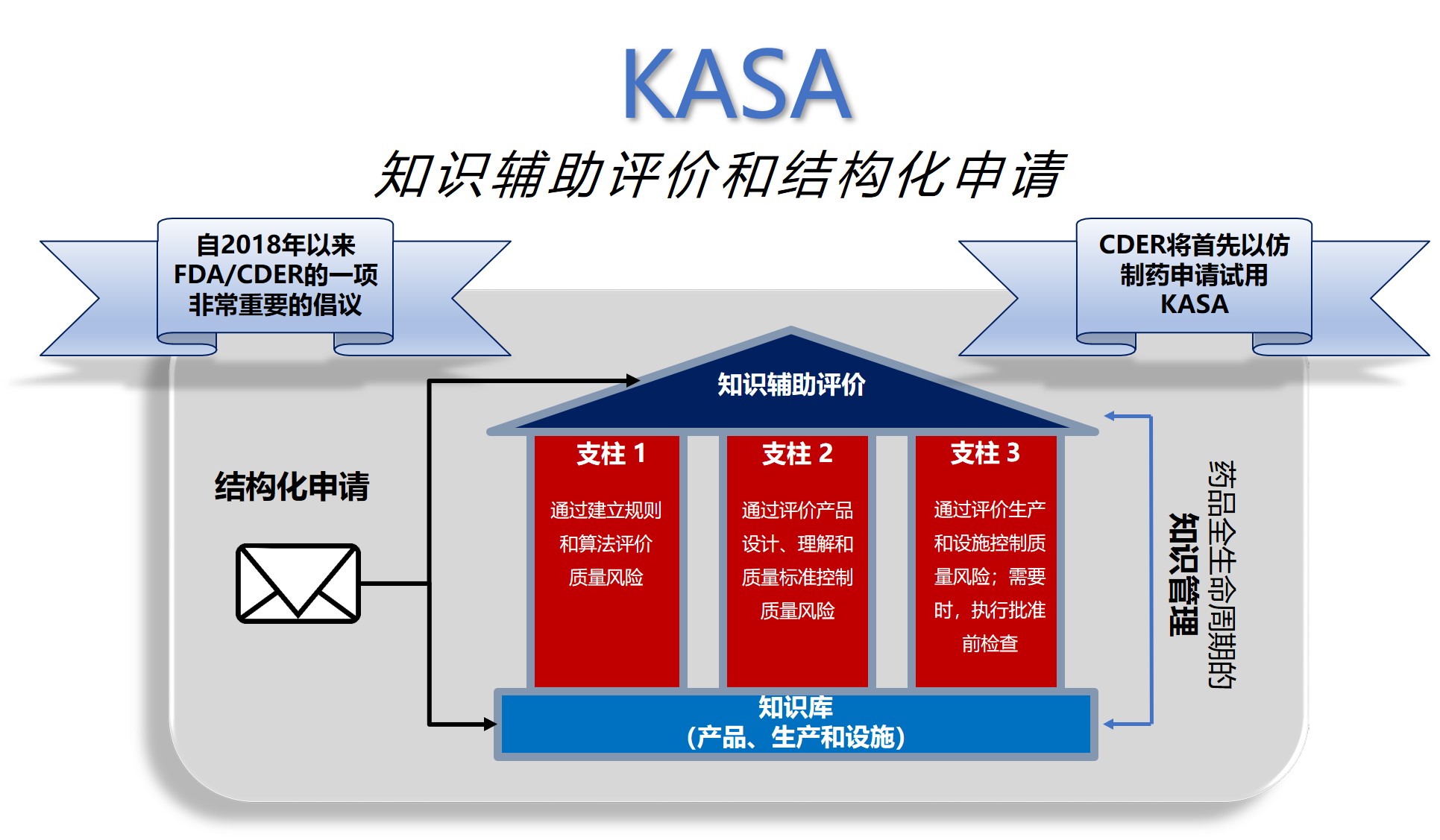
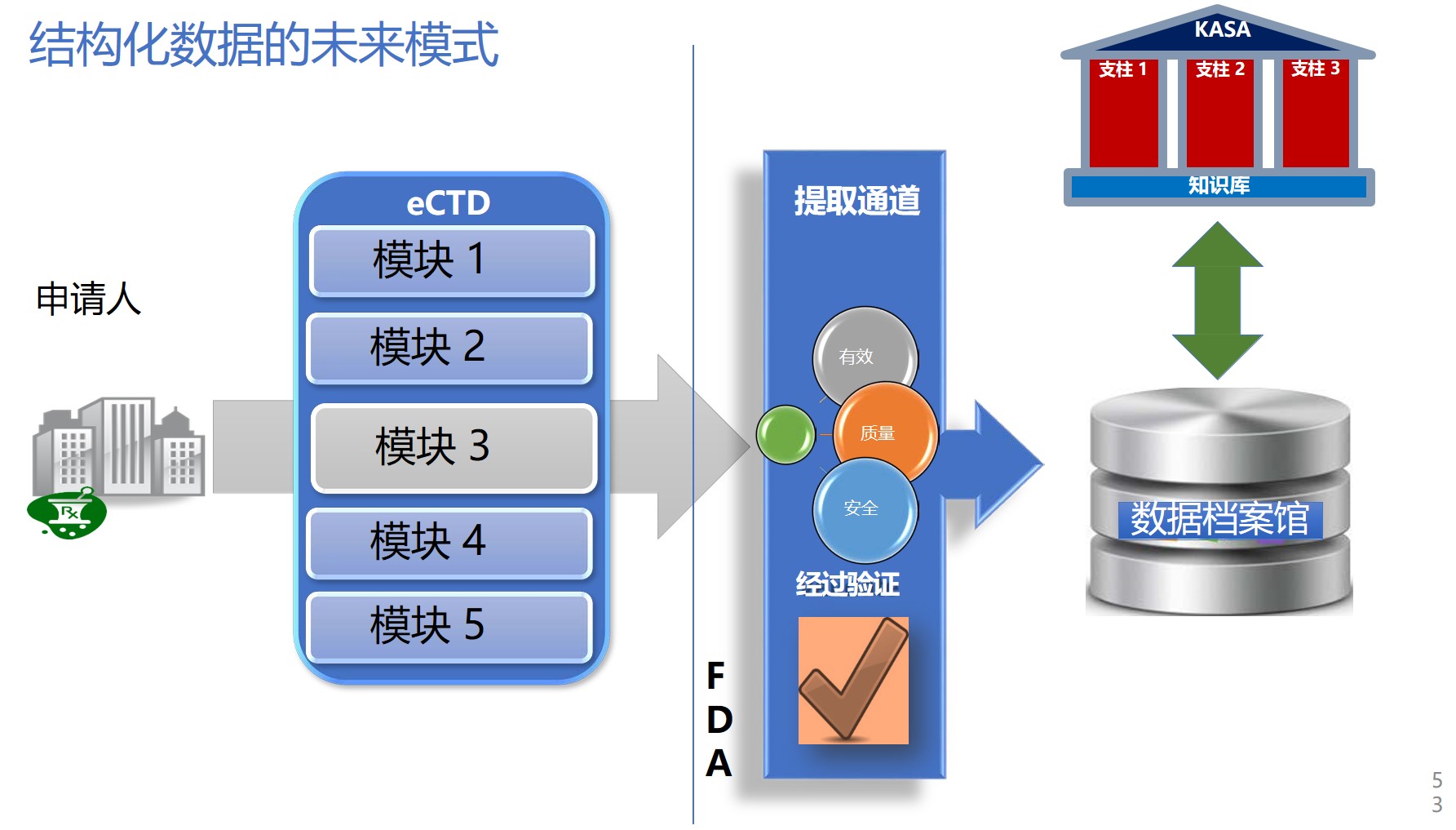
两周前在美国马里兰举行的PQRI大会上,FDA药品质量办公室副主任余煊强博士正式向业界宣布了面向21世纪的FDA药品评价(assessment*)新倡议:KASA(Knowledge-aided Assessment and Structured Application,之前翻译为知识辅助评价和结构化申请,余煊强博士建议翻译为智能评价和结构化申请更能反映其精髓)。该倡议由三个核心组成:eCTD 的“叙述式”关联;
余煊强博士介绍说,FDA已经成立了包括新药办公室、仿制药办公室、药品质量办公室和监管事务办公室在内的KASA项目小组。基于静态PDF的审评不满足高效、高质量的新评价追求。FDA将首先以仿制药评价,尤其是口服固体制剂 为出发点,并计划在2020年3月发布关于药品质量/CMC 项目的指南。
*编者注:Assessment 今后用于药品审批将译为“评价”,之前曾翻译为“评估”,但经过内部讨论,认为翻译为“评价”客观性更强,更能反映原意,故作此修改。【FDA 药品审批将不再使用“审评 review”一词】
支柱1:通过建立规则和算法评价质量风险
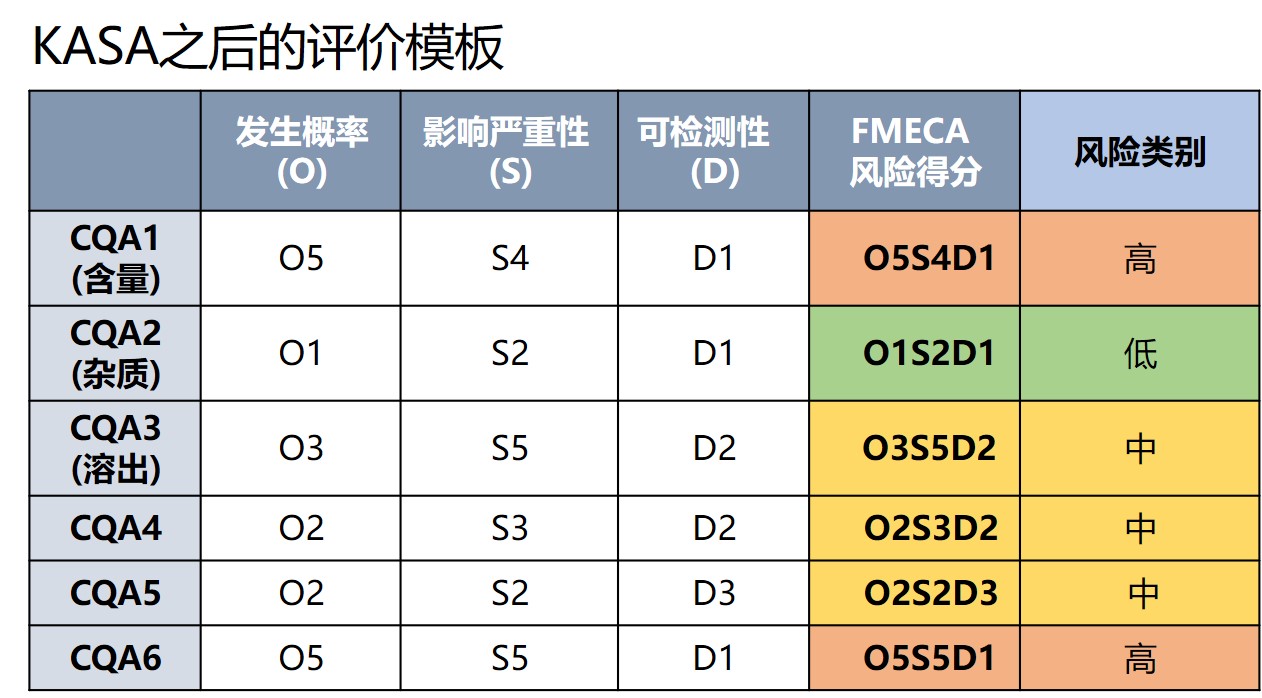
关键质量属性(CQA) 是评价的核心,FDA尝试通过失效模式、影响和关键性分析(Failure Modes, Effects and Criticality Analysis, FMECA) 来对CQA的风险排序,FDA通过历史评价数据和专家经验建立了知识库,提供了客观评价CQA出现失败的严重性、可观测性和发生频率的依据。并且可以根据信息的不断增加调整和提升算法,最终实现CQA风险评价的自动化。
支柱2:通过评价产品设计、理解以及质量标准控制风险
在对CQA自身风险评价之后,接下来评价CQA的风险是否得到了有效的控制,如果申请中没有充分的讨论控制,则为高风险,如果通过处方和工艺 设计减少了失效发生率,或者增加了放行前失效被检测到的概率,则可相应降低风险类别。
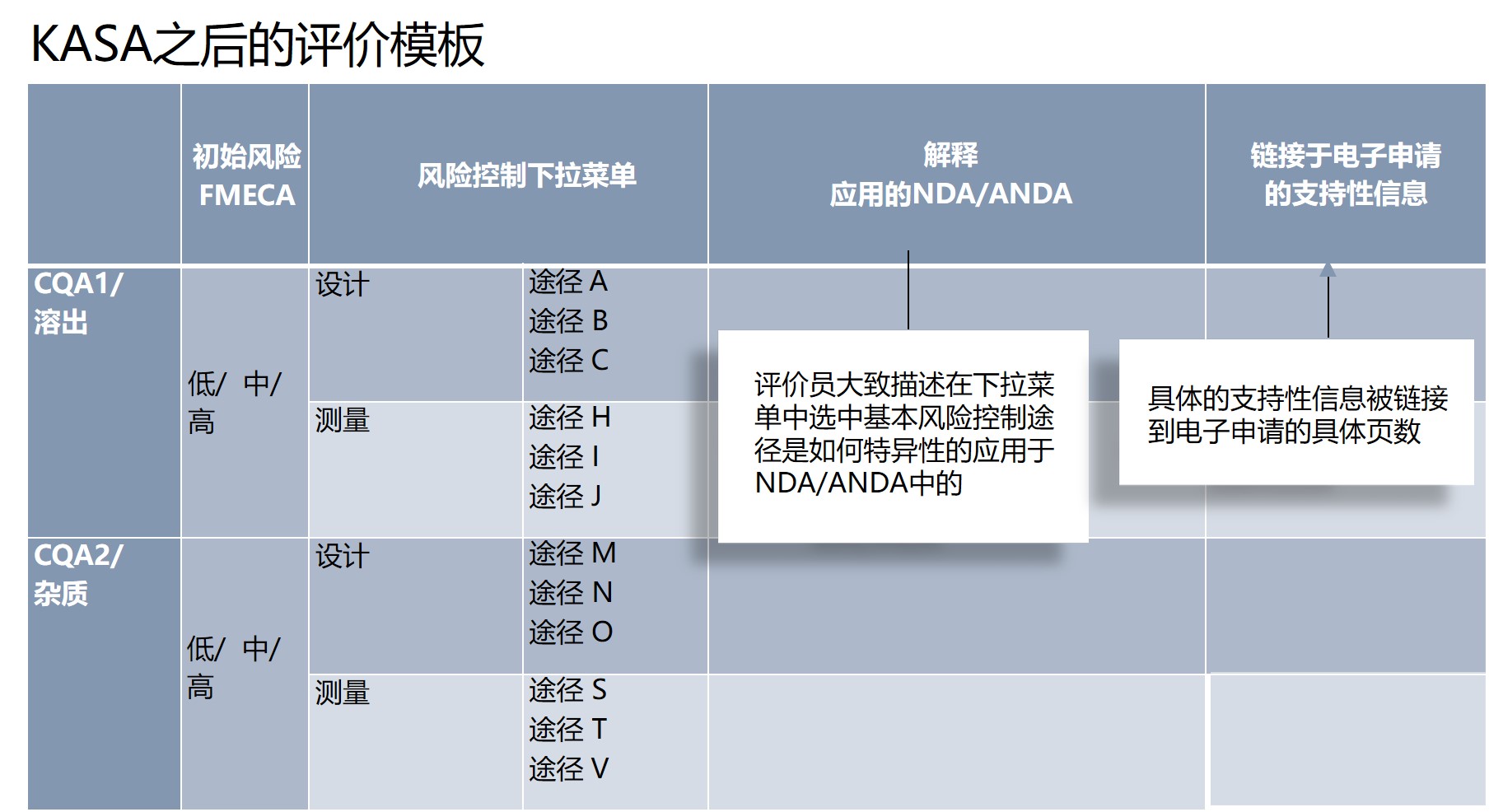
虽然CQA风险是通过计算得到的,但是内生的、客观的,而控制的方式却不是。目前“基于叙述性”的评价很大程度上依赖于审评员和专家的个人判断,也难以从之前的评价中提取参考依据。
KASA的优势是提供了横向比较的标准化模板,如,原研药、仿制药A和仿制药B,在相同的CQA(如,含量),内在风险都为高,但是原研药有多条设计和测量的控制途径,则风险为低,而仿制药A只有有限控制途径,风险为中,仿制药B缺乏控制途径,风险为高。
支柱3:通过评价生产和设施以及执行批准前检查 来控制风险
从生产角度看风险控制 ,评价的维度可能包括:
KASA通过支柱 3 的评价可以实现:
建立对持续风险控制和监管决策标准化的评价工具
识别需要批准前或批准后检查的设施
横向的比较产品和设施
基于对患者的风险来分配监管资源
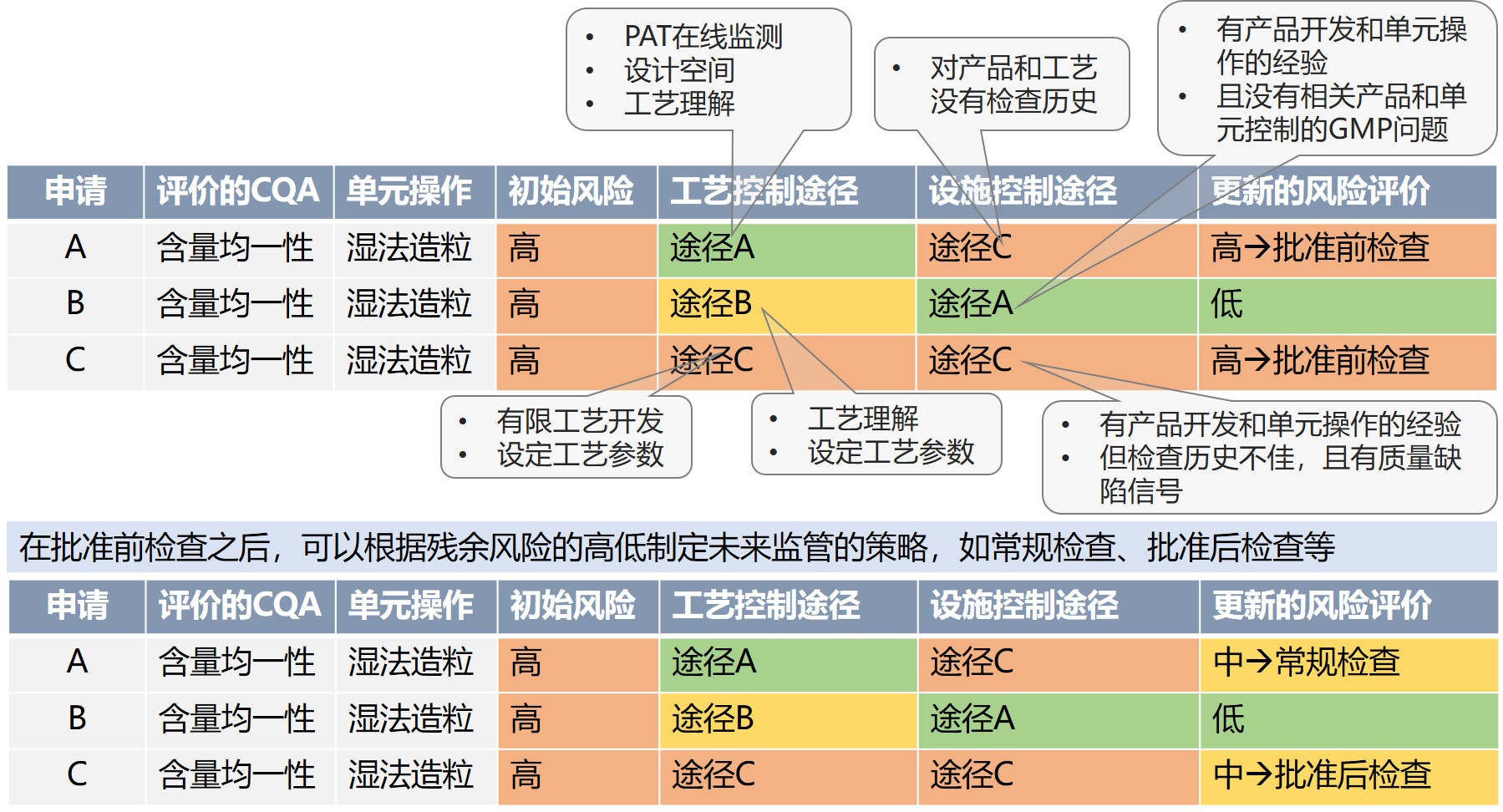
例如,对于A、B、C三件申请,关于CQA含量均一性 ,涉及到湿法造粒单元操作 ,初始风险(内生风险)为高,在控制策略 上三件申请水平有所差异,经过了良好的设计空间和工艺理解,且采用了PAT技术的A申请风险低,而工艺开发程度有限的C申请风险高,结合FDA过去的检查情况,A申请没有相关的检查历史,C申请检查历史不佳,而B申请的检查历史良好,因此最终对A和C进行批准前检查,而B企业不需要。根据检查的结果,确定还可能存在的风险高低(残余风险)来制定未来的监管策略。
问题:eCTD是否将被取代,过程是怎样的?
FDA在2018年提出KASA倡议后,广泛邀请产业界参与讨论和反馈,并于2018年9月召开了相应的专家顾问会(会议内容请见【FDA 知识辅助评估和结构化申请计划提上议程 2018/09/26】,会议视频请登录识林视频专区阅览)。余煊强博士在介绍中提到KASA和现有的eCTD模式的关系是业界关注的话题。比如eCTD中模块2 质量概要,是否已经包含了KASA所需要的信息,业界还需要准备好什么?
针对这些问题, KASA项目组代表指出,问题的核心是eCTD是静态的PDF数据,而且是“基于叙述性”的,需要手动的抓取药品评价所需要的真正“数据”,目前eCTD和KASA还是并行模式,但FDA已经开展了药品质量/CMC研究项目,旨在建立电子化药品质量和CMC数据的未来标准,以实现eCTD和KASA数据抓取和交换的自动化。
小结:KASA是连接过去和未来的知识管理系统
药品质量/CMC项目的实质是数据管理,着眼于注册申请中的数据的结构化,为KASA的实施提供数据交换的标准和工具,也为行业未来药品申请提供标准。KASA的实质是知识模型化、结构化、数据化及知识管理,不仅面向未来的申请,还可以从已有的申请和经验中提取知识,作为药品评价的参考,并提供产品、工艺和设施横向比较的技术途径。
制药行业是知识型产业,FDA已经做出了表率,良好的知识管理不仅能够提升监管和产业的效率,如,缩短药品评价时间,提高首轮批准率,而且能够使药品评价的标准更加透明和一致,节省了沟通和绕弯路中浪费的资源,最终更快为患者提供质量可比的产品。
作者:识林-枫® 版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
参考资料 识林资讯 FDA 知识辅助评估和结构化申请计划提上议程 2018/09/26 识林资讯:FDA 药品质量审评的新模式 – 识林现场报道 I 2019/04/10
岗位必读建议:
QA:确保口服固体制剂的生产过程符合本指南要求。 生产:遵循生产管理章节的指导,确保生产过程的合规性。 研发:在设计和选型设备时,参考本指南以确保设备符合生产需求。 临床:在产品实现和验证阶段,确保临床试验用药品的质量符合要求。 文件适用范围:
文件要点总结:
质量风险管理 :强调了质量风险管理在口服固体制剂生产中的重要性,包括原则和工具的应用。生产管理 :明确了生产过程中的关键控制项目,如批次管理和清场管理。设备要求 :规定了生产设备的设计、选型、校验、清洗、维护和使用记录。生产过程控制 :概述了工艺设计和过程单元操作的详细要求,包括配料、粉碎、混合等。物料管理 :强调了物料的接收、储存、分发、退库以及检验与放行的管理。以上仅为部分要点,请阅读原文,深入理解监管要求。