|
首页
>
资讯
>
国内药政每周导读:上海CAR-T监管规定,临床安全快速报告问答,“动物法则”药物注册,参比目录第56批
出自识林
国内药政每周导读:上海CAR-T监管规定,临床安全快速报告问答,“动物法则”药物注册,参比目录第56批
2022-07-25
【政策与监管综合】
7.15,安徽省局发布《关于落实“一改两为”持续优化审批服务的若干举措》
为支持本省医药创新,各省局纷纷发文,在法规允许的范围内为MAH、文号转让、委托生产等情形提供诸多便利。
安徽省局政策与众不同之处在于“创新现场检查方式”,提出“远程检查”,诸如:
- 多个品种同时变更生产场地的,可基于风险原则选择具有代表性的品种开展现场检查。
- 受托生产的生产线暂未通过GMP符合性检查的,受托企业可以在现场检查时,同步以拟受托生产品种开展GMP符合性检查。
- 我省持有人跨省委托生产已上市品种,现场检查和抽样工作由安徽省药监局协商受托方所在地省局开展。
- 持有A类药品生产许可证的企业因申报在研品种注册或品种转让而办理B类药品生产许可的,可基于风险原则实施远程检查。
【创新研发与临床】
7.19,CDE发布《急性髓细胞白血病新药临床研发技术指导原则(征求意见稿)》
急性髓细胞白血病(AML)是一种起源于髓系造血干/祖细胞的血液系统恶性疾病,各年龄阶段均可发病,尤其多发于老年患者。AML进展快、异质性强的特征,以及个性化精准治疗的临床要求,给新药临床研发带来了多重挑战。
本技术指导原则将从AML疾病特征出发,从人群定义、剂量探索、治疗策略探索、有效性终点、关键性注册研究设计、伴随诊断等方面探讨AML新药研发的关注点和技术要点,为致力于为AML新药研发的医药研发企业和研究者提供参考意见。
FDA于2020年8月发布《Acute Myeloid Leukemia: Developing Drugs and Biological Products for Treatment: Guidance for Industry》,可比较阅读。
【CMC与仿制药】
7.22,NMPA发布《仿制药参比制剂目录(第五十六批)》
参比制剂目录已公示六十批,正式发布五十六批。
识林用户可登录“中国参比制剂库”检索。
【注册,审评,审批】
7.19,CDE征求《药物临床试验期间安全性数据快速报告常见问答(2.0版)》意见
自2017年我国加入国际人用药品注册技术协调会(ICH)后,药物临床试验期间药物警戒监管工作驶入快车道。2018年,国家药品监督管理局药品审评中心(以下简称药审中心)建立了药物临床试验期间安全性快速报告的电子传输系统和数据库应用系统,并对外发布《药物临床试验期间安全性数据快速报告的标准和程序》,且于2019年4月发布了《药物临床试验期间安全性数据快速报告常见问答(1.0版)》。
2022年1月药审中心发布了关于药物临床试验期间个例安全性报告适用E2B(R3)区域实施指南的通知,指出为实施《个例安全性报告E2B(R3)区域实施指南》,申请人应及时完成系统配置,并按照区域实施指南要求实施E2B(R3),时间不得晚于2022年7月1日。
在此背景下,问答2.0征求意见,进一步指导临床期间药物警戒工作。
7.22,CDE发布《基于动物法则的药物注册技术指导原则(征求意见稿)》
某些用于治疗或预防由化学、生物、放射、核物质引起的严重威胁人体生命健康的疾病的药物开发对国家公共卫生战略部署和国家安全具有重大意义,但因开展人体有效性试验不符合伦理或现场试验不可行,无法按常规思路开展临床有效性试验获得人体有效性数据申报上市,这种情况下,药品监管部门基于充分的和良好对照的动物有效性试验数据及其它支持性数据,在确定该药物很可能带来临床获益时批准其上市,即基于“动物法则(Animal Rule)”的药物注册,如用于炭疽、鼠疫、核辐射等药物均可借鉴该策略进行上市有效性评价。
基于动物法则注册药物只因不符合伦理或现场试验不可行无法开展临床有效性评价,其他研究(如安全性等)需符合现有相关指导原则技术要求,故该指导原则对该类药物的一般研发思路仅进行简要概述,重点放在有效性评价方面。本指导原则主要分为8个章节,分别为概述、动物试验的一般要求、动物模型、充分的和良好对照的动物有效性试验、外推人体有效剂量、批准药物上市的附加条件、注释及参考文献以及附件。
【GMP与上市后监管】
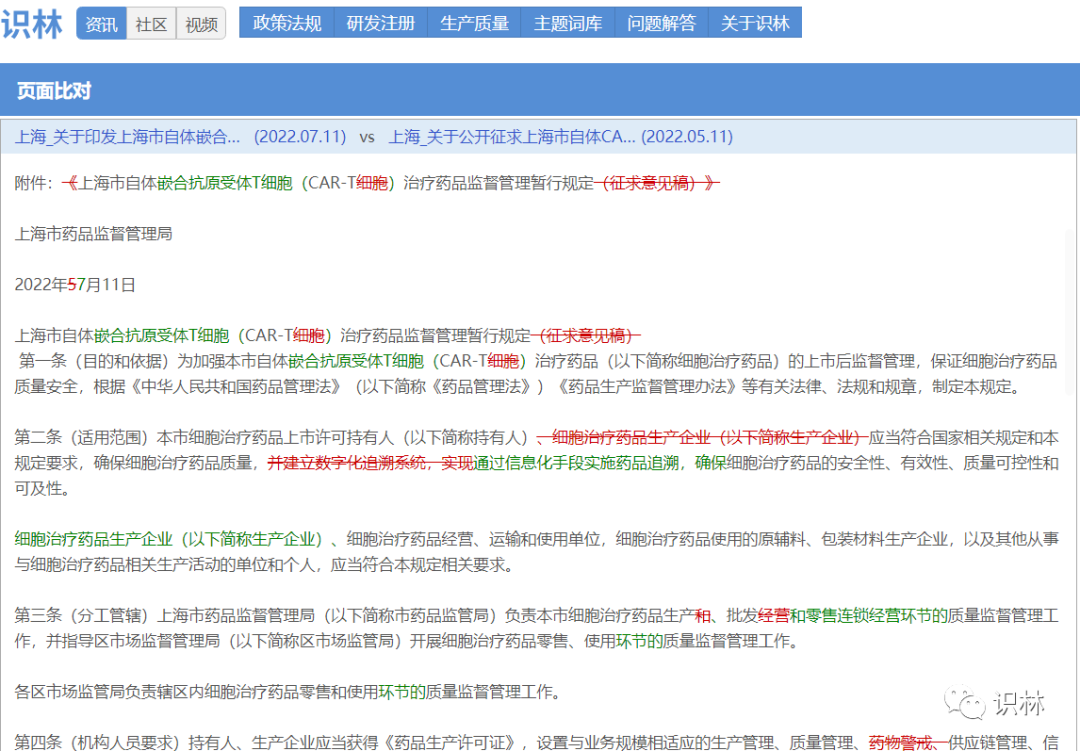
7.19,上海局发布《上海市自体嵌合抗原受体T细胞(CAR-T)治疗药品监督管理暂行规定》
征求意见仅2个月,国内第一份CAR-T系统性监管法规就正式发布。
通过识林“页面比对”制作花脸稿,可以看到一些值得关注的删改:
- 强调MAH的责任主体。第二条中,原本与MAH并列的“细胞治疗药品生产企业”换了位置,于是“应当符合国家相关规定”的就只有MAH,突出强调MAH作为药品责任主体。
- “建立数字化追溯系统”改为“通过信息化手段实施药品追溯”。措辞更灵活,强调目的而不是手段。
【中药相关】
7.20,浙江省人大发布《浙江省中医药条例》
条例是法的表现形式之一,具有法的效力。浙江省通过立法促进中医药发展。
其中与中药有关的内容包括药材保护,中药制剂调剂使用,中药饮片代煎等等。
对于中药创新,提出“知识产权保护机制”,并鼓励“形成具有自主知识产权的中药产品”。
此外,“医疗机构可以对其炮制的中药饮片、配制的中药制剂实施自主定价管理,对中药饮片的加成应当遵守国家有关规定。医疗机构应当对实施自主定价管理的中药饮片、中药制剂明码标价。”
7.20,广东省政府引发《广东省建设国家中医药综合改革示范区实施方案》
广东是中药大省,在这份行业促进政策中,与中药相关内容很多。
其中,与中药创新相关的有:“在中医药防治重大疑难、罕见疾病和新发突发传染病等基础和临床研究,创新中药及药源性肝肾毒性研究等方面,形成一批国内领先、具有核心竞争力的科研成果。支持深圳市创建中医药领域国家级创新平台,重点开展中医药防治脂肪肝、糖尿病及其并发症、肝癌、胃癌前病变等疾病的临床诊疗、科研转化和人才培养。”
且专门提及“中药国际化”,包括
- 深化粤港澳融合发展。推动广东省药品检验所建设中药国际标准权威研究机构,提升广东省港澳中药检定联合实验室的平台效应,促进粤港澳大湾区中药标准融合发展。
- 促进中医药海外发展。充分发挥粤澳合作中医药科技产业园“国家级中医药产品海外注册公共服务平台”“国家级科技企业孵化器”作用,推动中医药产品的海外注册及上市。
【新药批准和报产】
7.18-7.24,NMPA发布4个新药批准,CDE受理8个NDA
注:仅列出新药(包括改良型新药和生物类似药)的上市申请和批准上市信息。在“以临床价值为导向”的背景下,申请上市以及获批的品种,其适应症、临床研究策略、注册路径,都值得业界关注和分析。
识林-实木
识林®版权所有,未经许可不得转载
岗位必读建议: - PV(药物警戒):必读。需了解E2B(R3)区域实施的具体要求,确保个例安全性报告的合规性。
- QA(质量保证):必读。需监督药物警戒部门的报告流程,确保符合指南规定。
适用范围说明:
本文适用于所有在中国进行药品研发、生产和销售的企业,包括化学药、生物制品、疫苗和中药等药品类型,涉及创新药、仿制药、生物类似药和原料药等注册分类。由中国药品审评中心(CDE)发布。 文件要点总结: - 报告格式要求: 明确了E2B(R3)格式作为个例安全性报告的标准格式,强调了数据的标准化和一致性。
- 电子提交规定: 鼓励采用电子方式提交个例安全性报告,以提高报告的效率和准确性。
- 数据完整性与准确性: 强调了报告中数据的完整性和准确性,要求企业严格审核报告内容。
- 时效性要求: 规定了个例安全性报告的提交时限,要求企业在规定时间内完成报告的提交。
- 监管机构职责: 明确了CDE在个例安全性报告审核、评估和反馈中的职责和流程。
以上仅为部分要点,请阅读原文,深入理解监管要求。 岗位必读建议: - 临床研究(Clinical):熟悉快速报告标准和程序,确保临床试验期间的安全性数据及时、准确地报告。
- 药物警戒(PV):掌握个例安全性报告的电子传输技术文档要求,负责个例安全性报告的收集、评估和快速报告。
- 注册事务(Regulatory Affairs):了解报告程序和时限要求,确保注册过程中符合监管标准。
文件适用范围:
本文适用于化药、中药及生物制品的临床试验期间安全性数据快速报告。覆盖创新药、仿制药、生物类似药等注册分类,适用于中国境内的Biotech、大型药企、跨国药企等各类企业。 要点总结: - 快速报告责任:申请人负责临床试验期间非预期严重不良反应的快速报告。
- 严重不良反应定义:明确了导致死亡、危及生命等六种严重情形。
- 非预期不良反应判断:基于研究者手册等资料,与预期风险不同的不良反应应视为非预期。
- 报告时限和方式:规定了致死或危及生命的不良反应7天内报告,其他15天内报告的要求。
- 电子传输技术要求:ICSR应符合ICH E2B(R3)标准,通过Gateway或XML文件方式提交。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |