首页
>
资讯
>
国内药政每周导读:2024医保国谈启动,细胞治疗和肽类临床药理指南,临床急需药品进口优化,Q3C 适用,药典公示外源病毒检验法等
出自识林
国内药政每周导读:2024医保国谈启动,细胞治疗和肽类临床药理指南,临床急需药品进口优化,Q3C 适用,药典公示外源病毒检验法等
2024-07-01
上周国内药政导读
【创新与临床】
6.25,【CDE】关于公开征求《细胞治疗产品临床药理学研究技术指导原则(征求意见稿)》意见的通知
征求意见截至7月25日。
本指导原则旨在为细胞治疗 产品临床药理学 研究提供指导和建议,主要对细胞治疗产品临床药理学研究关键技术、研究内容及评价标准予以具体阐述。不同类型的细胞治疗产品体内过程也不尽相同,需根据不同细胞治疗产品的性质、作用机制、体内过程特点及目标适应症 适当调整研究内容和研究方法。
部分细胞治疗产品兼具细胞治疗和基因治疗产品的特性。本指导原则的目的不是对其监管属性或分类进行认定,而是基于现有认识,对细胞治疗产品开展临床药理学研究的若干技术问题提供建议和推荐。
6.25,【CDE】关于公开征求《肽类药物临床药理学研究技术指导原则(征求意见稿)》意见的通知
征求意见截至7月25日。
本指导原则中肽类药物主要指以肽类作为主要药效学基团的多肽,包括纯天然氨基酸的肽以及常规修饰氨基酸的肽、聚乙二醇修饰肽、脂肪链修饰肽等,而偶联其他基团而产生药理作用的药物(如放射性核素偶联药物)不在本指导原则中讨论。
肽类药物药代动力学(PK) 特征较复杂,开展肽类药物研究时应同时考虑小分子化合物和生物制品 的特点。
【CMC药学研究】
6.24,【CDE】关于发布《中药口服制剂生产过程质量控制研究技术指导原则(试行)》的通告(2024年第33号)
本文曾于2023年11月征求意见 。
本技术指导原则提出了中药口服制剂生产过程质量控制的研究策略和关注点,主要阐述了关键物料 质量属性和关键过程参数的确立、生产过程质量控制 方式的建立、生产过程质量风险的评估和方法验证 等内容,旨在为开展相关研究提供指导性意见。
鼓励药品上市许可持有人 对已上市中药开展生产过程质量控制研究,以产品质量稳定可控为目标,对生产工艺、设备进行优化,实现产品质量稳定可控和持续提高,并指导申请人 关注变更可能带来的变化,避免因工艺变更、设备更新换代出现“涨膏”等现象。
6.25,【NMPA】关于适用《Q3C(R9):杂质:残留溶剂的指导原则》国际人用药品注册技术协调会指导原则的公告(2024年第76号)
Q3C(R9)仅3.4 Analytical Procedures 小节修订,变化内容如下:
If only Class 3 solvents are present, a non-specific method such as loss on drying may be used, if the method is properly validated . The impact of solvent volatility on the test method should be considered in the validation.
若仅存在 3 类溶剂,可用非专属性的方法如干燥失重来检查(如果该方法经过适当验证)。验证时应考虑溶剂挥发性对检测方法的影响。
Validation of methods for residual solvents should conform to the current version of ICH guideline Q2 on Validation of Analytical Procedures.
残留溶剂的方法学验证应遵循当前版本的ICH指南Q2《分析方法验证》。
译文可参考Q3C(R8) CDE官网翻译
6.27,【CDE】关于公开征求《化学仿制药参比制剂目录(第八十四批)》(征求意见稿)意见的通知
截至目前,NMPA已发布参比制剂目录81批,CDE已公示84批 。
识林用户可登录“中国参比制剂库 ”查阅。
6.30,【药典会】发布多个公示,包括外源病毒,检定用动物,以及pH值检测等
公示的重要通则与指南包括:
可至识林中国药典数据库公示 查询近期公示。
【注册审评】
6.25,【NMPA】公开征求《国家药监局关于进一步优化临床急需境外已上市药品审评审批有关事项的公告》意见
征求意见截至7月24日。
NMPA提供多个措施鼓励临床急需新药进口,包括:
6.30,CDE 药品审评报告和说明书更新,其中包括下列国产新药
注:仅列出国产创新药和改良型新药 ,多受理号仅展示其中1个,供参考。企业用户可至识林“CDE 药品审评报告 ”按发布时间查阅最新的上市药品信息。
受理号
药品名称
药品类型
注册分类
企业名称
承办日期
CXSS2300017
特瑞普利单抗注射液
治疗用生物制品
2.2
上海君实
2023-04-11
CXSS2200015
注射用曲妥珠单抗
治疗用生物制品
3.3
正大天晴
2022-02-17
CXSS2200004
葎草花粉点刺液
治疗用生物制品
1
浙江我武
2022-01-13
CXSS2101078
白桦花粉点刺液
治疗用生物制品
1
浙江我武
2021-12-27
CXSS2101079
黄花蒿花粉点刺液
治疗用生物制品
1
浙江我武
2021-12-27
CXSS2200015
注射用曲妥珠单抗
治疗用生物制品
3.3
正大天晴
2022-02-17
CXSS2200004
葎草花粉点刺液
治疗用生物制品
1
浙江我武
2022-01-13
CXSS2101078
白桦花粉点刺液
治疗用生物制品
1
浙江我武
2021-12-27
CXSS2101079
黄花蒿花粉点刺液
治疗用生物制品
1
浙江我武
2021-12-27
CXHS2101031
依鲁奥克片
化药
1
齐鲁
2021-07-24
CXHS2101023
盐酸安舒法辛缓释 片
化药
1
山东绿叶
2021-06-11
【政策监管综合】
6.28,【国家医疗保障局】关于公布《2024年国家基本医疗保险、工伤保险和生育保险药品目录调整工作方案》及申报指南等文件的公告
2024医保国谈持续推进。
在往年经验做法的基础上,今年对《工作方案》进行了小幅调整。主要包括以下三方面:
一是申报条件方面。按规则对药品获批和修改适应症的时间要求进行了顺延,2019年1月1日以后获批上市或修改适应症的药品可以提出申报。
二是调出品种的范围方面。将近3年未向医保定点医药机构供应的常规目录药品,以及未按协议约定保障市场供应的谈判药品列为重点考虑的情形,强化供应保障管理。
三是强化专家监督管理。明确专家参与规则和遴选标准条件,加强对参与专家的专业培训和指导,提高评审测算的科学性、规范性。建立健全专家公正履职承诺、保密管理、对外宣传等规定。
今年的工作程序仍分为准备、申报、专家评审、谈判、公布结果5个阶段。7月1日正式启动申报,争取11月份完成谈判并公布结果。
【新药批准和报产】
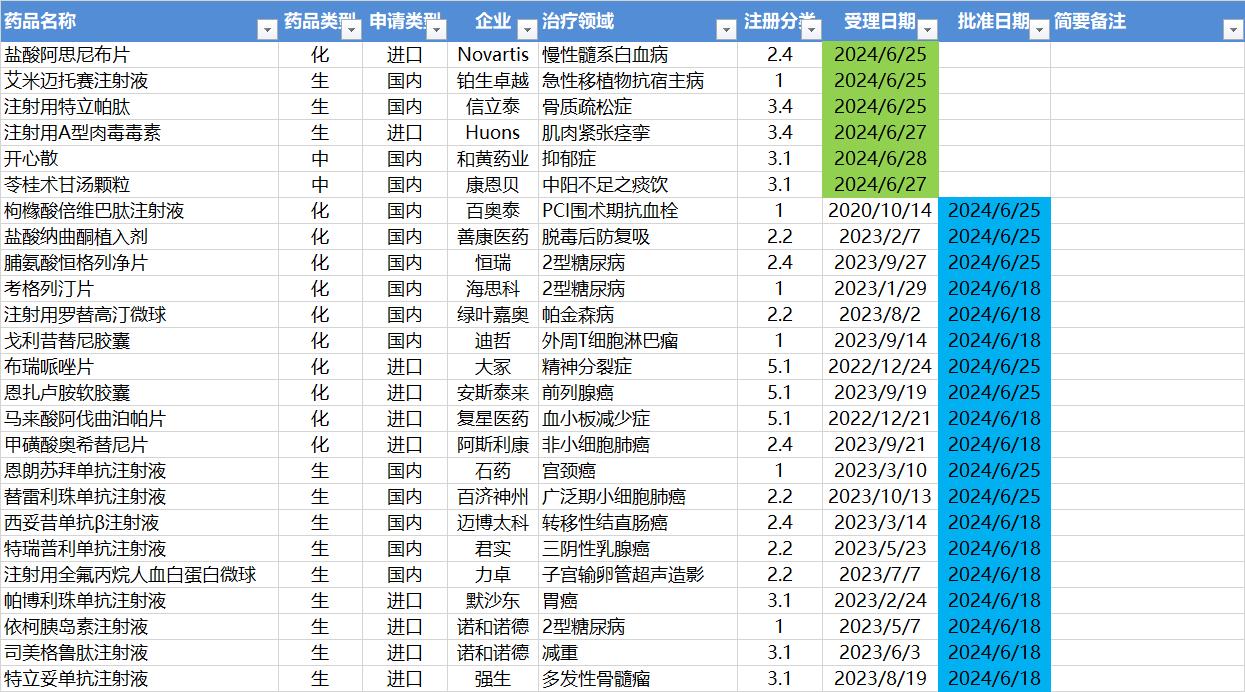
6.24-6.30,NMPA发布19个新药批准,CDE受理6个NDA
注:仅列出新药(包括改良型新药 )的上市申请和批准上市信息。在“以临床价值为导向”的背景下,申请上市以及获批的品种,其适应症 、临床研究 策略、注册路径,都值得业界关注和分析。
识林® 版权所有,未经许可不得转载。
岗位必读建议:
注册专员 :了解参比制剂目录的最新动态,为仿制药注册策略提供依据。研发人员 :关注未通过审议品种目录,避免在研发过程中选择不适宜的参比制剂。质量管理(QA) :确保公司仿制药研发与生产过程符合参比制剂的要求。文件适用范围:
文件要点总结:
征求意见公示 :对第八十四批化学仿制药参比制剂目录进行公示,并征求意见。反馈要求 :反馈意见需提供充分依据和论证材料,并加盖单位公章。公示期限 :公示期限为2024年6月27日至7月10日,共计10个工作日。未通过审议品种 :详细列出了未通过审议的品种及其原因,如不具有参比制剂地位、未提供充分的安全有效性数据等。放射性药物特殊性 :特别指出放射性药物的参比制剂主要用于明确研发目标和基本要求。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA(质量保证):应熟悉分析方法验证的全过程,包括验证研究、生命周期管理、报告范围确定等,以确保质量控制流程的合规性。 R&D(研发):需要理解分析方法验证的具体要求,以科学的方法开发和优化分析过程。 QAC(质量控制):必须掌握分析方法验证的测试和评估方法,确保日常质量控制的准确性和精确性。 注册部门:应了解分析方法验证的法规要求,以便在药品注册文件中准确呈现相关信息。 文件适用范围:
文件要点总结:
分析方法验证目的 :验证分析程序适用于预定目的,如确保分析结果的准确性和可靠性。验证研究设计 :应设计验证研究以提供足够证据,证明分析程序满足其目标,包括性能特征和相关标准。生命周期中的验证 :在分析程序的生命周期中,可能需要部分或全部重新验证,以应对变更。报告范围 :分析程序的报告范围应包括所有可报告结果的值,确保精度和准确性。稳健性评估 :评估分析程序在预期操作环境中的适用性,包括对分析程序参数的故意变化。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA:确保残留溶剂的分析程序和报告水平符合ICH Q3C(R9)指南要求。 R&D:在药物合成过程中,选择和使用溶剂时应考虑本指南的分类和限制。 Production:在生产过程中控制和监测残留溶剂,确保符合PDE标准。 Analytical Chemists:使用适当的分析方法验证残留溶剂的存在,并考虑溶剂挥发性对测试方法的影响。 文件适用范围:
文件要点总结:
分析程序修订:对于仅存在3类溶剂的情况,可以使用如干燥失重这样的非专属性方法,前提是该方法经过了适当的验证,并考虑了溶剂挥发性对测试方法的影响。 方法验证:残留溶剂的分析方法验证应遵循ICH指南Q2《分析方法验证》的当前版本。 残留溶剂分类:根据风险评估,将残留溶剂分为三类,其中1类溶剂应避免使用,2类溶剂应限制使用,而3类溶剂具有较低的毒性潜力。 暴露限值建立方法:为残留溶剂建立的允许日暴露量(PDE)基于最相关的动物研究的无观察到效果水平(NOEL)或最低观察到效果水平(LOEL)。 报告残留溶剂水平:供应商应提供关于辅料或药物成分中残留溶剂含量的信息,以帮助制药产品制造商满足本指南的标准。 以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA:应仔细研读修订草案,确保公司产品pH值测定方法符合最新标准。 研发:需关注pH值测定方法的更新,以保证研发过程中的准确性和合规性。 生产:了解新标准,确保生产过程中pH值测定的准确性。 文件适用范围:
文件要点总结:
修订公示 :《中国药典》0631 pH值测定法的修订草案已公示,征求社会各界意见。公示期限 :公示期为60天,自2024年6月28日至2024年8月28日。反馈要求 :若有异议,需在线反馈并附相关说明、实验数据和联系方式,个人来函需签名。默认同意 :公示期满未回复意见即视为对公示标准草案无异议。联系信息 :提供了联系人、电话、电子邮箱和通信地址,以供反馈和咨询。以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位建议:
QA(质量保证):确保生物制品生产和检定过程中使用的实验动物符合新修订的质量控制标准。 生物制品研发:在实验动物选择和使用上遵循新标准,保证研发过程的合规性。 生物制品生产:按照新标准对实验动物进行质量控制,确保生产过程的合规性。 适用范围说明:
文件要点总结:
公示目的 :修订《中国药典》中关于生物制品生产及检定用实验动物的质量控制标准,以提高标准的科学性、合理性和适用性。公示期限 :公示期为60天,自2024年6月28日至2024年8月28日。反馈机制 :鼓励社会各界提出异议,需在线反馈并附相关说明、实验数据和联系方式。个人或单位反馈需加盖公章或签名。默认同意 :公示期满未回复意见即视为对公示标准草案无异议。联系信息 :提供了联系人、电话、电子邮箱和通信地址,以便社会各界进行沟通和反馈。以上仅为部分要点,请阅读原文,深入理解监管要求。