继美国 FDA 于 4 月 1 日要求所有雷尼替丁产品撤市后,欧盟也于 5 月 1 日发布通告,因可能含低水平的亚硝胺杂质,暂停所有欧盟境内的雷尼替丁产品销售和使用。近年来,亚硝胺杂质问题一波未平一波又起,一直是监管机构、工业界和公众的关注焦点。今天我们编译了一篇发表在 Pharmaceutical Technology 上的文章,期望能够为在药物开发过程早期识别遗传毒性杂质,将遗传毒性杂质调节在 API 或制剂的可接受水平,以及在开发和/或销售过程后期避免负面产品监管措施(包括昂贵的召回)提供方向。
过去几年中,控制 API 中的诱变和/或遗传杂质一直是 FDA 和其他监管机构关注的重点。在探究这些缺陷的根源并找出避免这些缺陷的积极措施之前,重要的是了解诱变杂质和遗传毒性杂质之间的关系。长期以来,这两个术语都被监管机构和制药行业互换使用,但是,ICH M7(R1)[4]在标题和内容中都特别提到了“诱变”杂质,而不是一般所说的“遗传毒性杂质”。因此,在开始讨论之前,弄清楚与诱变杂质和遗传毒性杂质之间的关系是有帮助的。根据 ICH M7(R1) 中的定义,遗传毒性是“一个广义术语,指的是遗传物质中的任何有害变化,与引起变化的机制无关”。而《全球化学品统一分类和标签系统》(GHS)第五修订版第 3 章对诱变和遗传毒性物质有以下定义:
“3.5.1.3 突变一词既适用于可能在表型水平上显示的可被遗传的基因变化,也适用于已知的基础 DNA 修饰(包括例如特定碱基对的变化和染色体易位)。术语诱变剂和致突变剂将被用于在细胞和/或生物体中引起突变增加的试剂。”
“3.5.1.4 更笼统的术语遗传毒性和遗传毒性作用适用于改变 DNA 的结构、信息含量或分离的试剂或过程,包括那些通过干扰正常复制过程而导致 DNA 损伤,或以非生理的方式(临时)改变其复制的试剂或过程。遗传毒性检测结果通常作为诱变作用的指标。”
因此,诱变性意味着诱导细胞或生物体遗传物质的数量或结构发生永久性的可传递的变化,而所有遗传毒性作用并不一定与突变相关。因此,所有诱变剂均具有遗传毒性,但并非所有遗传毒性物质均具有诱变性。当在 FDA 和其他监管机构的出版物和缺陷中提及遗传毒性杂质时,包括诱变剂以及其它可通过不同途径造成 DNA 损伤的杂质。本文中,除非特别需要将物质称为致突变物质,否则术语“遗传毒性杂质”将被用于指代致突变性以及其它遗传毒性杂质。
文章讨论的重点是提交到 FDA 的申报资料,尤其是针对 API 已获得 FDA 批准的仿制药的申报,包括 505(j) 简化新药申请(ANDA)和 505(b)(2) 新药申请(NDA)。但文中的大多数原则也适用于新化学实体以及其他遵循 ICH 指南的国家和地区监管。
讨论
遗传毒性杂质可能会因 API 的制造、API 的降解或在某些情况下从辅料中带到制剂中。API 和制剂中遗传毒性杂质的来源通常是 API 的制造过程,包括起始物料和试剂。在 API 合成中使用的试剂通常具有很高的反应活性,遗传毒性杂质可能是制造过程中的残余试剂、化学转化的副产物或 API 的后续降解/相互作用所致。尽管很少见,但遗传毒性杂质有时会由于 API 与辅料相互作用在成品制剂中形成。本文重点介绍 API 制造过程中可能产生的遗传毒性杂质。
未能认识到形成具有遗传毒性杂质的可能性会严重影响 NDA 或 ANDA 的获批。通常与 505(b)(1) 申请相关的新化学实体,申办人会对合成路径进行评估,并在研究用新药申请(IND)流程的早期阶段解决可能存在和有理由存在的杂质,包括具有遗传毒性可能性的杂质。在最佳情况下,在提交 NDA 之前,申办人会与 FDA 分享控制策略并达成共识。然而,对于 505(j) 和某些 505(b)(2) 申请,这些申请所涉及的 API 已经在FDA已批准的药物中使用,在这种情况下,对与 API 制造过程相关的风险的详细了解通常会处于次要位置。无论 API 是否存在于FDA已批准的产品中,基于新的合成途径、起始物料的来源、试剂的来源和所使用的溶剂,都可能存在与遗传毒性杂质有关的风险。这在 505(b)(1) NDA 的生命周期管理中也可能是一个问题。至关重要的是,必须对 API 的制造过程进行适当的评估,并对起始物料和试剂的来源和控制进行适当的审查,以确保在 FDA 审评或生命周期管理期间杂质谱中不会出现意外。
大家通常会提出一个问题,即,在 API 生产工艺中从哪里开始确定可能的遗传毒性杂质。最好的起点是认识图1中提供的与遗传毒性有关的结构性预警。
起始物料。起始物料的选择在 API 合成中至关重要,原因有很多,其中之一就是起始物料对 API 杂质谱的影响。不应在工艺的下游确定起始物料,因为这可能会使清除源自起始物料的杂质变得困难。另外,还需要了解起始物料的生产工艺,并且需要很好地理解工艺中用到的任何试剂或中间产物或副产物中是否存在对遗传毒性杂质具有结构性预警作用的物质。如果在被定义为监管起始物料(RSM)的后期中间体中存在潜在遗传毒性杂质,则 API 制造商可能会承担着艰巨的任务:在 RSM 和最终 API 中添加重要控制措施或说服 FDA 这些杂质可以通过工艺进行有效清除并且不会对 API 或药品造成风险。两种选择都既昂贵又费时,并且可能导致批准延迟。
反应副产物。如果制造过程中副反应的任何副产物具有遗传毒性的结构预警,则需要对其进行评估,以确定它们是否在后续反应中被完全消耗或在整个制造过程中持续存在。如果需要,应在适当的步骤中包括对这些副产物的控制。控制杂质的最佳位置为接近其来源的位置。但是,如果副产品保留在最终 API 中,则可能需要在来源点以及 API 处添加控制。
降解物。如果潜在遗传毒性物质或杂质除了在制造过程中产生的以外,还可能是 API 的降解物,则需要对 API 中以及成品制剂中的杂质进行控制,以保证在制剂中的最大日剂量(MDD)。
控制策略。一旦在 API 制造过程中确定了可能的遗传毒性杂质,就需要制定与控制它们相关的策略。该策略可以从了解基于 MDD 的可接受杂质摄入量以及 ICH M7(R1) 表2中提供的治疗时间开始。根据药物是长期使用还是短期使用,可接受的摄入量可能存在显着差异。对于根据适应症具有不同 MDD 的药物,应使用最保守的 MDD 来确定毒理学关注阈值(TTC)。在确定之后,应开发出足够灵敏的分析方法,以检测和定量所建议水平的潜在遗传毒性杂质。对于上游产生的潜在遗传毒性杂质,可以进行加样和清除研究,以确保这些杂质的含量低于最终 API 中 TTC 含量的30%。控制策略也可以基于对杂质性质以及测量或预测的清除因子的科学理解。
如果杂质在 API 制造过程的下游产生并根据加样和清除或清除因子研究持续存在于 API 中,并且/或是已知的降解物,则应控制 API 和制剂中的杂质。TTC 以及加样和清除研究可以证明在中间体或最终 API 中要控制的遗传毒性杂质的限量。ICH M7(R1)指南提供了对遗传毒性杂质进行分类以及确定安全水平的方法。其它指南,例如关于遗传毒性测试的 ICH S2(R1),如果根据定量的结构活性关系鉴别出遗传毒性杂质,则可以确定细菌和哺乳动物的体外和体内测试方案。例外情况是目标化合物与 API 本身具有相同的结构预警。在这种情况下,可以将其视为非诱变杂质,并根据 ICH Q3A(R2) 确认或鉴别阈值进行控制。
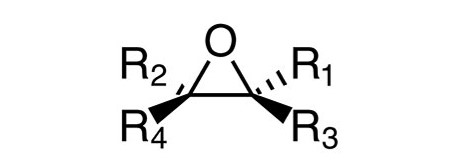
环氧化物。环氧化物(如图2所示;其中R1-4为烷基、芳基或 H)是可以与 DNA 相互作用的高反应性化合物,被认为对基因毒性具有结构性预警。环氧化合物可能作为反应的中间体形成,也可能在合成途径涉及烯烃的生成或使用以及与氧化剂(例如过氧酸)一起形成的情况下形成。环氧化合物通常在强酸和醇存在下反应,生成非遗传毒性的乙二醇。
烷基卤化物。烷基卤化物(如图3所示;其中R =烷基、芳基或H,X =碘、氟、氯或溴化物和其它良好的离去基团)在 API 的合成中被广泛用作烷基化剂。由于烷基卤化物被过量使用,因此它们大多在烷基化反应结束时作为残留物存在。烷基卤化物无处不在,通常用于许多 API 生产工艺中。下面的例子是一些已经明确鉴别出这些杂质的药物。它们大多是高反应性的(例如,烷基化剂),并且是已知的遗传毒性和致癌化合物。
亚硝胺。亚硝胺(如图5所示;其中R1-2为烷基或芳基)是众所周知的遗传毒性和致癌化学物质,广泛存在于环境和食物链中。当仲胺(API 结构的一部分或制造过程中的试剂)在低pH环境中暴露于亚硝化剂(如亚硝酸钠)时,亚硝胺通常在 API 或制剂中形成。当试剂和溶剂中存在仲胺杂质时,也会形成亚硝胺。因此,如果在 API 的制造过程中使用有机或无机亚硝酸盐,申办人应根据在过程中可能使用或形成的胺或酰胺来评估亚硝胺的可能性。
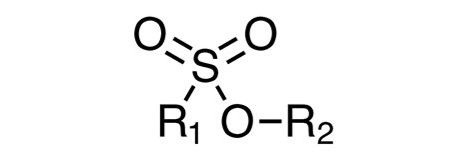
磺酸酯。磺酸酯(图6;其中R1 =烷基或芳基;其中R2 =烷基)。磺酸酯是在API制造过程中形成的最常见的遗传毒性杂质之一。其背后的原因是烷基磺酸酯、烷基磺酸、醇或烷基卤化物的前体是有机反应中使用最广泛的物质。许多 API 是烷基磺酸的盐,如甲磺酸酯、甲苯磺酸酯、甲苯磺酸酯和三氟甲磺酸盐,导致 API 中存在化学计量比的烷基磺酸共轭碱。同样,烷基磺酸还用于官能团的保护和脱保护,并且在一些反应中还用作酸催化剂。像甲醇、乙醇和异丙醇这样的醇是 API 生产过程中常用的溶剂。同样,可以与烷基磺酸反应生成潜在的具有遗传毒性的磺酸酯的烷基卤化物本身被认为是具有遗传毒性的结构预警,也是 API 生产中常用的试剂。磺酸酯的形成可以根据反应过程中存在的水或游离碱的含量进行控制。
遗传毒性杂质需要在药物开发过程的早期预期和鉴别。API 制造过程中潜在遗传毒性杂质的较晚发现可能会导致由于制造过程的中断而引发的申报延迟。监管机构在审评过程中要求评估潜在的遗传毒性杂质可能导致监管机构批准的延误和市场份额的损失。如果在上市后进行鉴别,则可能会导致药品供应中断,在某些情况下还会导致药品短缺。在 API 制造过程中识别出任何结构性预警,以及定量结构活性关系(QSAR)模型阳性确认,表明需要按照 ICH M7 (R1)的要求进行细菌反向突变测定以进行确认和/或创建控制策略。申办人对于可能因选择的 API 制造工艺而产生的潜在遗传毒性结构预警的认识,以及有关如何控制潜在遗传毒性杂质的早期计划,将是朝着缩短审评周期迈出的重要一步,而这反过来可能是快速批准的通道。毕竟,正如本杰明·富兰克林曾经说过的那样,“防范胜于补救(An ounce of prevention is worth a pound of cure)”。
A. Srinivasan, “Proactive Evaluation of Possible Genotoxic Impurities During the Early Stages of Drug Development," Pharmaceutical Technology APIs, Excipients, and Manufacturing Supplement (October 2019).
参考资料
1. N. Filiz, “Content of the Dossier–The Top Deficiencies Identified in Dossiers,” Certification of Substances Department, European Directorate for the Quality of Medicines, May 2018.
2. O. Diego, et. al., J Pharm Pharm Sci., 17(2), pp 169–186 (April 2014).
3. H. Liao, “Impurity Case Studies: Control of Potential Genotoxic Impurities in DMF,” Generic Drug Forum, April 4, 2019.
4. ICH, ICH Harmonised Guideline–Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk, M7(R1), March 31, 2017.
5. United Nations, Globally Harmonized System of Classification and labelling of Chemicals (GHS), Fifth Revised Edition (United Nations, New York and Geneva, 2013).
6. FDA, “FDA Updates and Press Announcements on Angiotensin II Receptor Blocker (ARB) Recalls (Valsartan, Losartan, and Irbesartan),” updated July 24, 2019.
7. FDA, “FDA Provides Update on Its Ongoing Investigation into ARB Drug Products; Reports on Finding of a New Nitrosamine Impurity in Certain Lots of Losartan and Product Recall,” Press Release, March 1, 2019.
8. FDA, “Statement Alerting Patients and Health Care Professionals of NDMA Found in Samples of Ranitidine,” Sept. 13, 2019.
9. A. Giordani et al., Eur J Pharm Sci., 43 (1–2), pp 1–15 (March 2011).
10. G. Szekely, et al., Chem. Revs., 115 (16), pp. 8182−8229 (August 2015).
11. J. Wisler and K.A. Black, “Assessment of Genotoxic Impurities in Small Molecule Drug Candidates,” Amgen Inc., Northern California SOT Meeting, May 6, 2010.
12. S. Thotla, et. al., Ind. Eng. Chem. Res., 46 (25), pp 8371-8379 (May 2007).
13. A. Srinivasan, A., “Nitrosamines–How to Address these Unwelcome Guests in The Pharmaceutical World,” Contract Pharma, May 27, 2019.
14. A. Teasdale, et. al., Org. Process Res. Dev., 17(2), pp 221-230 (February 2013).
15. ICH, Guidance on Genotoxicity Testing And Data Interpretation For Pharmaceuticals Intended For Human Use S2(R1), (ICH, November, 9, 2011).
16. C. Limban, et. al., Toxicol Rep., 5, pp. 943–953 (August 2018).
17. J. Abolghasem and H. Parsa, “Genotoxic Impurities in Pharmaceuticals, Toxicity and Drug Testing,” William Acree, IntechOpen, DOI:10.5772/24030, February 10, 2012.
18. L.S. Gold et al., “The Carcinogenic Potency Database (CPDB),” UC Berkeley Lab., September 1, 2011.
19. D. Elder et al., J Pharm Biomed Anal., 54 (5), pp 900–910 (April 2011).
20. R. N. Loeppky, “Nitrosamine and N-Nitroso Compound Chemistry and Biochemistry: Advances and Perspectives,” ACS Symposium Series, Vol. 553, Chapter 1, pp. 1–18, March 28, 1994.
21. P.N. Gillatt et. al., Food Chem Toxicol., 1984, 22(4), pp 269–274 (April 1984).
22. A. Teasdale et. al., Org Process Res Devel 14(4), pp 999–1007 (March 2010).
23. J. D. Wichard, Food Chem Toxicol., 106(B), pp 595–599 (August 2017).
要点总结: ICH M7(R1)指南旨在为药物中的DNA反应性(基因毒性)杂质提供评估和控制框架,以限制潜在致癌风险。指南强调了在药物合成和降解过程中产生的基因毒性杂质的识别、分类、鉴定和控制。特别指出,对于已知的基因毒性致癌物,应基于致癌潜力和线性外推计算特定的可接受摄入量(AI),或使用国际监管机构已发布的值。对于表现出非线性剂量-反应关系或具有实际阈值的化合物,可基于无观察到效应水平(NOEL)和不确定性因子来计算每日允许摄入量(PDE)。指南还提供了基于治疗持续时间的不同可接受摄入量的调整方法,并讨论了多种控制策略,包括过程控制和定期检测。此外,指南还涉及了对上市产品的考虑,包括对药物合成、制剂变更和临床使用变更的评估。