|
首页
>
资讯
>
FDA仿制药办公室发布首份年度报告
出自识林
2016-04-14 FDA
仿制药为所有美国公众提供了更多的医疗保健机会。
对于FDA药品审评与研究中心(CDER)仿制药办公室(OGD)来说,2015年是重要的一年。这是我们在极大地扩展办公室的范围和构架成为“超级办公室”后的第一个完整年度。这一改变使得办公室具有更加重要的地位,并拥有更多的工作人员处理日益增加的工作量,提高我们推进在美国的仿制药的安全性和可及性。
想想看:2014年,仿制药为美国卫生系统节约了约2540亿美元 — FDA将继续努力推进仿制药的使用,以帮助改善公众健康。
我们职责的增加和工作范围的扩张发生在一个关键时刻。2012年,一项新的法律称为仿制药使用者付费法案(GDUFA)为FDA授权额外的资金用于仿制药申请审评、设施检查和其它监管行动。但伴随这些额外资金而来的是FDA承诺达成各种目标。这些目标随着GDUFA立法在一份文件中做了阐述,这份文件是由FDA和工业界谈判达成并由国会颁布的。额外的资金帮助FDA有效地处理成千上万件新的仿制药产品申请,并减少审评批准仿制药所需的时间。
我们有望满足所有目标。今天,为帮助公众了解我们的进展,OGD发布了我们的首份年度报告。这份年度报告充满了我们工作的详细记录,我们的工作是以对申请更高效的审评来改善仿制药计划,通过发展所需要的科学来帮助仿制药企业证明他们的产品与品牌产品同样安全有效。
年报中的亮点是,报告指出,2015年标志着有史以来FDA做出的仿制药批准和暂时批准最高的一年 — 总共超过700件。去年12月,自仿制药计划启动以来,我们达到单月批准和暂时批准最高数量(99件)。
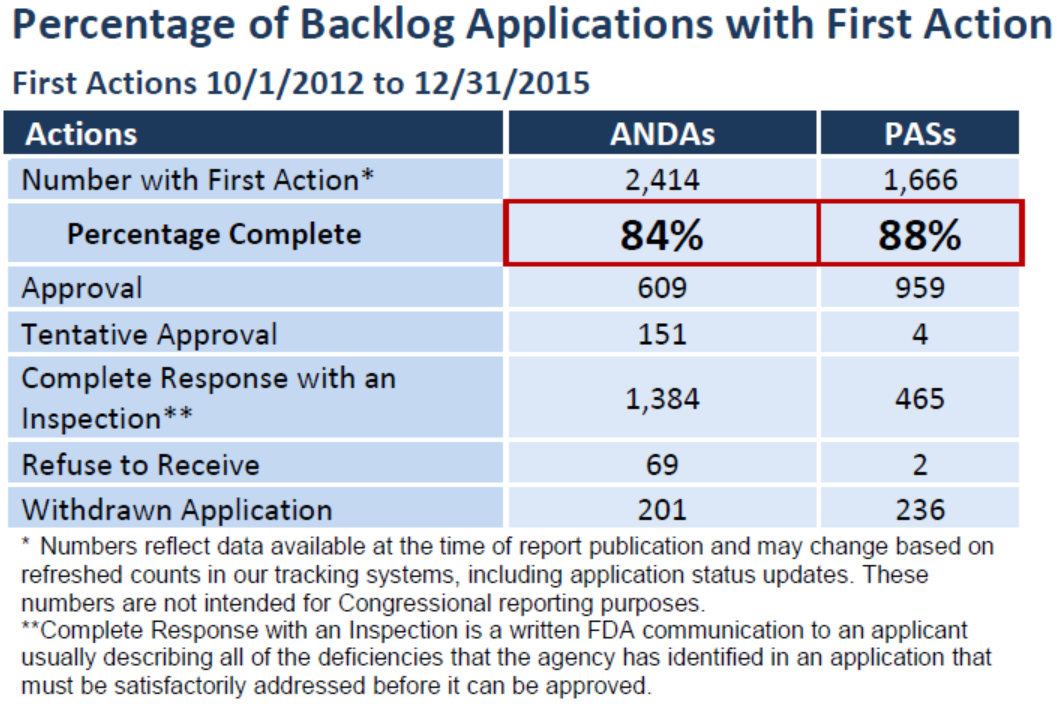
GDUFA另外一项重要承诺是到2017年对90%的“积压”首次采取行动,这些积压申请是在2012年10月1日GDUFA之前未决的申请。我们拥有2,866件简化新药申请(ANDA)和1,873件之前已获批申请的补充申请(PAS),但到2015年底,我们对84%的ANDA和88%的PAS完成了首次行动 — 已经接近设定的2017年完成90%的目标!我们已经批准90个“首仿药”,意味着在2015年我们为90个品牌药增加了新的节约费用的仿制药替代。
尽管我们已经取得了进步,我们的前路仍然漫长。但我们不期望完全靠我们自己获得成功。实现为公众工作的目标需要来自公众,包括工业界、学术界、立法者和其他利益攸关者的共同努力。
作为我们与利益攸关者的愿景保持一致的努力的一部分,我们将于5月20日举行公开会议,征求对我们的监管科学举措的宝贵意见,帮助我们指明前进的方向。我们邀请所有利益攸关者通过向我们的公共卷宗提交您的想法和意见参与并贡献您的力量。
我们建议您阅读我们的年度报告,并参加我们的年会。随着我们不断的努力和强大的公众投入,我们相信2016年以及未来将与2015年一样成功。
作者:Kathleen “Cook” Uhl 医师,FDA药品审评与研究中心仿制药办公室主任
翻译自 FDA Voice - "2015: An Important Year for Advancing Generic Drugs at FDA" , 2016/04/13
2015仿制药办公室年度报告概览
FDA仿制药计划
仿制药占美国处方量的88%,2005年到2014年间,仿制药为美国医疗体系节约了1.68万亿美元。其结果就是FDA的仿制药计划变得越来越资源不足。FDA仿制药计划离不开FDA其它相关办公室与仿制药办公室的共同努力。
仿制药办公室(OGD)
仿制药办公室直接向药品审评与研究中心(CDER)主任报告,由一个直属办公室和四个下属办公室组成。2015年OGD招聘125名新员工,实现了GDUFA招聘目标。分别介绍了直属办公室和4个下属办公室的架构和主要职责。
GDUFA:使仿制药计划走向成功
OGD的成功很大程度上得益于GDUFA,根据GDUFA,企业同意每年支付3亿美元,作为交换FDA承诺绩效目标,具体目标包含在FDA与企业谈判商定的“仿制药使用者付费法案计划绩效目标和程序协议”中(“GDUFA承诺函”)。GDUFA I期整体是五年计划,于2017年终止。FDA与企业就下一个GDUFA协议的谈判正在进行中,将特别关注FDA的绩效目标。
2015年底,FDA已经达到或超过“GDUFA承诺函”中列出的所有当前可衡量的绩效目标。
对GDUFA之前(“积压”)申请的行动
国会担心的主要问题之一是,不久之前,等待FDA采取行动的ANDA积压严重。积压申请被定义为2012年10月1日GDUFA之前摆在FDA面前的的未决申请。FDA在承诺函中承诺到2017财年末对90%的积压申请采取首次行动。
截止2012年10月1日,积压包括2,866件ANDA和1,873件之前已获批申请的补充申请(PAS),但到2015年12月31日,FDA对84%的ANDA和88%的PAS完成了首次行动。
受控函
受控函是FDA为了帮助企业开发申请回答的产品研发问题。在2014年10月GDUFA目标日期开始之前FDA已经有很多受控函积压。FDA目前已经基本消除受控函积压。2015年OGD创纪录的处理了2,065封受控函。
改进业务流程
作为实现GDUFA目标的策略之一,OGD继续精简现有流程以建立更高效的体系,落实书面流程例如标准操作程序和政策与程序手册(MAPPs),并向业务信息学办公室提供输入,他们面向CDER内部的审评人员推出了新的信息平台。
ANDA立卷积压已基本消除。立卷是FDA评估药品申请人递交的申请是否足够完整以允许FDA开展实质性审评的过程。2014年8月有1100多件申请还没有经过初始立卷审查。今天已经没有积压,立卷是实时进行的。OGD对2015财年递交具有GDUFA目标日期的99%的ANDA在60天之内发布立卷决定,立卷决定平均在大约40天之内做出并传达给企业。
指南和标准
OGD发布具体产品的建议(或称“生物等效性指南”)以促进有效的ANDA立卷审评、仿制药上市前研发,并精确的追溯OGD在这方面的行动。在决定发布哪些指南文件方面,OGD考虑制剂的复杂性、在作用部位准确测量其生物利用度的能力、证明生物等效性的科学方法和同类药物之前的经验和知识。OGD还通过分析收到的请求和分析100个最畅销药品和其它广泛使用药品的清单考量此类指南的需求水平。
现在有超过1,300份具体产品BE指南公布在网上。2015年,OGD发布124份新的和48份修订的具体产品BE指南文件。2015年发布的许多指南涉及复杂剂型,例如吸入粉剂、鼻腔喷雾、局部产品和眼科产品。
2015年,OGD发布以下关于仿制药的指南文件:
2015年,OGD发布以下MAPPs:
增进与企业和利益攸关者的沟通
2015年,OGD与仿制药企业和其他利益攸关者通过以下各种平台沟通:
- 行业监管教育(REdI)仿制药论坛,每年与CDER小企业帮助组(SBIA)合作举办,向行业提供机会与FDA主题专家讨论仿制药批准程序。
- OGD新沟通举措网络研讨会,详细介绍了ANDA申请人可以从OGD新沟通承诺中期待什么,例如实时回复审评人员问题并及时从监管项目管理人员处获得更新。
- “行业指南:仿制药研发相关的受控函”教学网络研讨会提供了对向FDA递交受控函请求与仿制药研发相关的信息过程相关的额外洞察。
- 关于专利和专营权、药物主文件和其它行业感兴趣的更新信息通过SBIA简报(SBIA Chronicles)和邮件订阅(Small Biz Buzz)共享。
批准和其它监管行动
2015年OGD取得580件批准和146件暂时批准总共超过700件批准的成绩,成为有史以来批准量最高的一年。包括12月共99件完全批准和暂时批准,自仿制药计划启动以来单月批准和暂时批准量最高的月份。
FDA认为首仿药是公共卫生的重点工作,优先审评这些递交。在年报附录中,OGD列出了值得关注的已获批首仿药清单。
2015 GDUFA监管科学计划
作为GDUFA实施的一部分,FDA同意与公众、产业界和学术界磋商,制定针对仿制药研究的监管科学举措年度清单。到目前为止,GDUFA已经在研究项目中资助3490万美元。监管科学的成果将在所有产品类别中为FDA评估仿制药等效性和企业高效开发新的仿制产品提供新的工具,从而增加对安全有效仿制药的获取。
新的和持续的GDUFA研究资金分布在整个GDUFA监管科学优先领域:
1. 仿制药上市后评估
2. 复杂药品的等效性
3. 局部作用产品的等效性
4. 治疗等效性评估和标准
5. 跨领域计算和分析工具
2015年GDUFA监管科学计划最重要的成果来自两个在癫痫患者中的批准后BE研究。在患者中的BE研究结果与用于原始批准的健康受试者BE研究结果完全一致。这些研究结果为支持抗癫痫药物(AED)成功的仿制药替代提供了证据。
参考资料
岗位必读建议: - 注册(RA):必须熟悉本指南,以便在提交ANDA时能够提供符合FDA要求的标签草案,并确保最终印刷标签(FPL)与批准的标签一致。
- 研发(R&D):应了解本指南,以确保在ANDA开发过程中,标签草案的内容和格式符合FDA的规定。
- 质量保证(QA):需要了解本指南,以监督ANDA标签的合规性,并确保标签的最终版本与批准的草案一致。
文件适用范围:
本文适用于美国FDA监管下的化学仿制药(ANDA),适用于Biotech、大型药企、跨国药企等提交ANDA的各类企业。 文件要点总结: - 标签草案接受性:FDA接受ANDA申请中的标签草案,而不需要最终印刷标签(FPL),只要草案标签的缺陷仅限于编辑或类似的小问题。
- ANDA批准条件:ANDA可以基于草案标签批准,条件是草案标签满足以下推荐标准。
- 标签内容与格式:ANDA申请必须包含与上市药品相同标签的草案或最终印刷标签的副本,并以电子格式提交标签内容。
- 标签审查:OGD将审查标签草案,以确保其在安全、格式、颜色、字体大小和风格等方面符合要求。
- 最终印刷标签责任:基于标签草案批准ANDA的申请人需确保最终印刷标签与批准的标签文本完全一致。
以上仅为部分要点,请阅读原文,深入理解监管要求。 岗位必读建议: - 注册专员(注册):熟悉FDA对通用药品开发相关控制通信的指导,以便在提交申请时遵循正确的程序和格式。
- 研发人员(研发):了解控制通信的定义和适用范围,确保在药品开发过程中及时获取FDA的反馈。
- 质量管理(QA):掌握控制通信的提交和审查流程,保证企业提交的通信符合FDA要求。
文件适用范围: 本文适用于美国FDA对通用药品制造商及相关行业在药品开发过程中提交的控制通信的指导。涵盖化学药品的仿制药,由美国FDA发布,适用于Biotech、大型药企、跨国药企等。 文件要点总结: 控制通信定义:明确了控制通信是针对通用药品开发特定要素的信息请求,不包括公民请愿书等。 控制通信范围:详述了哪些问题属于控制通信范畴,包括悬而未决的公民请愿问题、仍在考虑中的问题等。 提交控制通信:提供了提交控制通信的方法、内容要求,以及特定类型控制通信的额外建议。 控制通信审查:解释了不同FDA部门如何审查控制通信,以及推荐合适的审查学科。 FDA的通信:描述了FDA如何与提交控制通信的请求者进行沟通,包括确认接受、回应和目标日期。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |