首页
>
资讯
>
FDA 总结 2019 财年警告信四大趋势
出自识林
2019-09-24
美国 FDA 生产质量办公室副主任 Rick Friedman 于 9 月 16 日在华盛顿举行的 PDA/FDA 联席监管会议上,对 2019 财年最新 GMP 违规导致的警告信发布情况做了通报,并通过最近一些警告信 来举例说明了检查员发现的一些缺陷项。
本财年到目前为止生产质量办公室已发布 89 封警告信,主要缺陷项包括 API 的亚硝胺检测、无菌设施设计不良、对水系统的控制不足导致微生物污染以及重新包装商掩盖原始 API 制造商身份导致供应链中 API 可追溯性不足等。
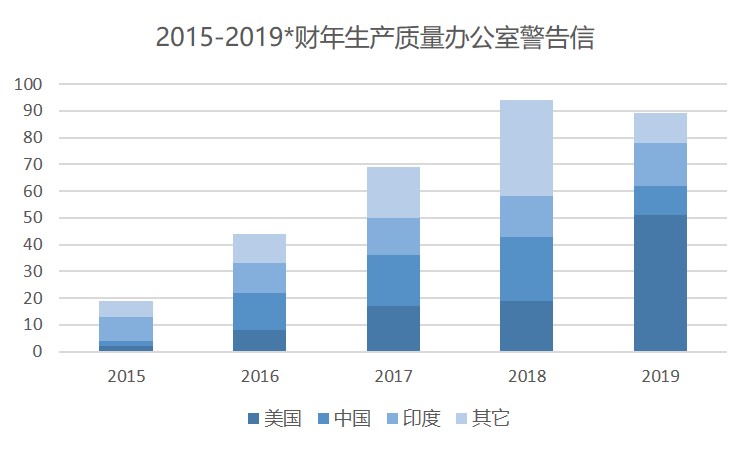
FDA 的数字显示,由 FDA 生产质量办公室发出的警告信从 2015 财年的 19 封开始,每年都在稳定增长。2018 财年 FDA 发布了 94 封 GMP 警告信。发送给美国制药商的警告信数量已超过去年。到目前为止,FDA 已向美国企业发出 51 封警告信,而去年仅有 19 封。
不包括配药方面的警告信
Friedman 表示,今年在警告信中看到的四个“显著”趋势是:
供应链缺陷,例如 API 杂质问题以及再包装商对于原始制造商的标识缺乏透明度;
水系统检测不足;
无菌处理设施设计不充分;
数据可靠性 问题。
杂质问题
Friedman 表示,API 杂质检测不充分仍是 FDA 关注的主要问题。缬沙坦危机和降压药召回的数量不断增加,已经促使 FDA 宣布更加关注对 API 供应商的检查,以确定是否有必要的控制措施来检测原料药中的亚硝胺。他表示有两封警告信表明 FDA 对于供应商在生产原料药时未检测到致癌杂质的担忧。
FDA 于 8 月 8 日向回收溶剂供应商 Lantech 公司发出警告信,该企业的回收溶剂含有潜在诱变杂质,可能导致了终产品的污染。并于今年 6 月份将该公司列入进口禁令清单中。【缬沙坦危机新进展:FDA 瞄准回收溶剂供应商 2019/08/20】 此前 FDA 于 6 月份曾向 Aurobindo 公司发布警告信 指出企业未能报告导致原料药污染的溶剂中的杂质。Friedman 表示,沙坦类药品中的潜在致癌物质是全球监管机构都尤为关注的问题,因为其与大多数其它杂质不同,“有可能在非常低的水平上造成伤害”。
从这些警告信中所吸取的教训是,供应商必须生成所有 API 的完整杂质分布图,以确保识别出潜在的致癌物质,而生成这些分布图通常需要使用多种检测方法。
再包装商缺乏透明度
今年出现的另一趋势是 API 的再包装商无法维持供应链中原始 API 制造商的可追溯性。这种担忧从 FDA 向 API 再包装商发出的三封 GMP 警告信中可一窥端倪。FDA 药品审评与研究中心(CDER)主任 Janet Woodcock 表示,这种透明度的缺乏是对公共卫生的真正威胁,必须得到尽快解决。【FDA 连发警告信加强对原料药再包装商的监管 2019/07/25】
水系统检测不足
Friedman 表示,2019 年还针对微生物控制不力发出了多封警告信,例如对用于生产非无菌药物的制药用水系统监督不严。他表示,有大量因水污染“而导致的召回事件”。
他举了 3 月 20 日发给北卡罗莱纳州阿什维尔的顺势治疗 药物生产商 King Bio 的警告信为例,该公司未能解决水系统受污染的问题。FDA 在警告信中指出,“多年来,用作药物成分的水的检验结果以及顺势疗法成品药的结果重复出现微生物限度超标。检验显示存在极高水平的微生物污染,包括过多而无法计数(TNTC)结果,并且发现药品中存在严重的机会致病菌”。Friedman 表示,“我们与公司 CEO 通了多次电话”,公司最终召回了所有水基产品。【FDA 因 GMP 违规对顺势治疗产品公司发布警告信 2019/04/04】
无菌处理设施设计不足
Friedman 表示,另一经常看到的违规行为是无菌处理设施的设计不当,有 19 封发送给企业的警告信是由于这方面的违规,同时也是造成药品短缺 的主要原因。
1 月 4 日发给伊利诺伊州迪凯特市 Akorn 公司的警告信指出,一名检查员发现无菌生产线干预措施中存在多处缺陷,例如胶塞多余,西林瓶堵塞和掉落,以及在干预过程中阻断了首次空气,物料流动不良和烟雾研究不足等。【Akorn 因根源调查不力再收 FDA 警告信 2019/07/12】
数据可靠性问题仍然存在
Friedman 表示,FDA 继续将数据可靠性问题以及计算机系统完整性的缺乏视为主要根本原因。该问题的一个例子是太多人共享计算机管理员权限。他表示,“公司必须格外注意”确保这些权限“不能出现在实验室中”。拥有管理员权限的人员必须在实验室之外。Friedman 表示,PDA 正在制定有关数据可靠性的技术报告,FDA 还计划很快发布有关数据可靠性的问答指南更新。
另外,Friedman 指出,其它方面的警告信趋势还包括超标(OOS) 调查不充分。他敦促企业查阅 Barr 法院判决 ,该判决涉及如何记录和处理 OOS 结果,并阅读 FDA 2006 年的“药品生产中 OOS 检测结果的调查”行业指南 。美国政府诉 Barr 制药公司案由美国新泽西地区联邦地区法院于 1993 年作出裁决。(【FDA对Wolin法官对美国政府诉Barr制药公司案法院裁决中所包含的GMP议题解读的归纳】 ,【法院裁决书】 )
警告信中显示出的其它问题包括冻干周期问题,由于使用手动或过时的生产工艺而导致的生产过程不可靠,清洁不充分以及甘油成分检测不足等。
作者:识林-Aspen® 版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
参考资料 Joanne S. Eglovitch. FDA: Key FY 2019 Warning Letter Trend Is Inadequate Testing For API Impurities 识林资讯:缬沙坦危机新进展:FDA 瞄准回收溶剂供应商 2019/08/20 识林资讯:FDA 连发警告信加强对原料药再包装商的监管 2019/07/25 识林资讯:FDA 因 GMP 违规对顺势治疗产品公司发布警告信 2019/04/04 识林资讯:Akorn 因根源调查不力再收 FDA 警告信 2019/07/12 Barr制药公司案法院裁决书 FDA对Wolin法官对美国政府诉Barr制药公司案法院裁决中所包含的GMP议题解读的归纳
岗位必读建议:
QA(质量保证):负责监督OOS调查流程,确保调查的全面性和合规性。 QC实验室人员:直接参与OOS结果的初步评估和实验室调查,需熟悉相关操作和记录要求。 生产部门:参与全面OOS调查,评估生产过程中可能导致OOS结果的因素。 研发部门:在产品设计和开发阶段,需考虑OOS结果对产品质量的潜在影响。 文件适用范围:
文件要点总结:
OOS结果定义与重要性 :明确OOS结果包括超出药品规格或接受标准的所有检测结果,强调OOS结果调查的必要性。实验室调查责任 :规定分析师和实验室主管在OOS结果初步评估中的责任和行动。全面OOS调查 :详述当初步评估未能确定实验室错误时,如何进行包括生产过程审查和额外实验室测试的全面OOS调查。报告测试结果 :讨论了在OOS调查中适当和不适当使用平均值和异常值测试的情况。调查结论 :指导如何根据调查结果解释、做出批放行决定,并在必要时提交现场警报报告。以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。 QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。