首页
>
资讯
>
GRx Biosims 大会简报:FDA 的仿制药申报、审评与检查
出自识林
GRx Biosims 大会简报:FDA 的仿制药申报、审评与检查
2025-11-07
2025年10月27日-29日,AAM(美国普享药协会)GRx+Biosims会议 在美国马里兰州贝塞斯达举行。Lachman事务所资深仿制药政策顾问Bob Pollock参与会议,重点关注仿制药 申报、审评缺陷 以及检查和警告信等议题。即使美国政府停摆(10月1日开始,已是美国史上最长) 、裁员导致士气低迷以及其他种种政策限制,FDA多位官员还是出席了会议。
以下内容摘要自Pollock的分享,供我国出海仿制药企参考FDA仿制药监管最新动态。
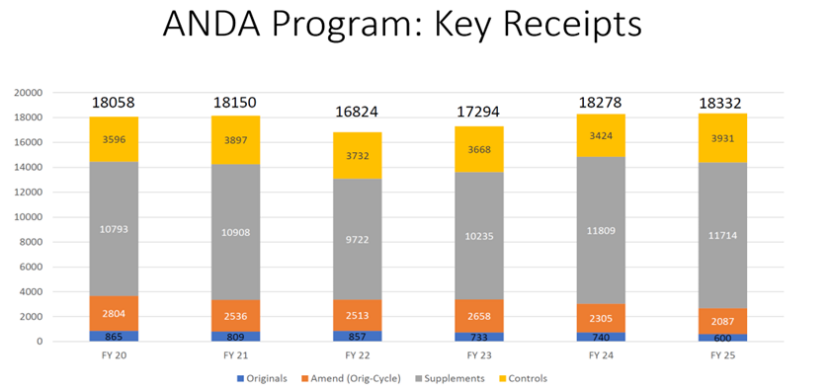
新的ANDA申请量逐年下降
会议首日,FDA仿制药办公室(OGD)主任Iilun Murphy介绍2025财年仿制药申报数据。数据显示,OGD在2025财年仅收到600份新的ANDA (仿制药申请),较2024财年的740份大幅下降。
尽管ANDA提交数量减少,但OGD批准了689份新的ANDA,仅略低于2024财年的694份。财年内ANDA批准和暂时批准总数为939份。
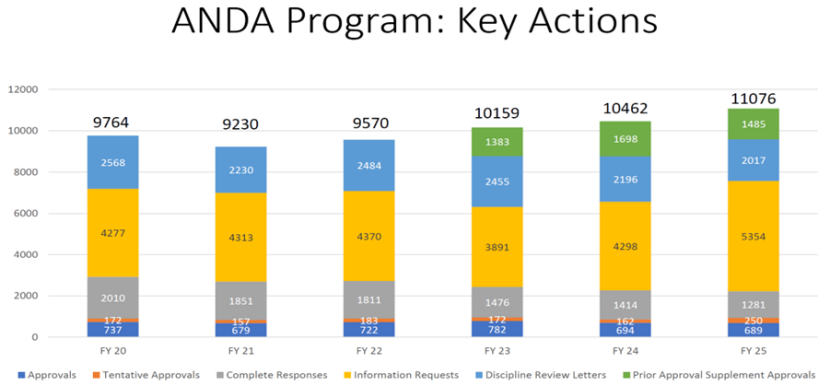
数量并不代表工作量。如果考虑到其他ANDA提交、补充、修正和受控通信的总量,OGD的工作量在过去六个财年中最高。此外,关键项目行动(包括ANDA批准、暂时批准、完全回应函 、信息索要、学科审评函 和补充申请 批准)在2025财年也达到过去六年最高水平。
16%的拒收可能仅因为356h表格,损失近9万美元
会议期间,Bob Pollock受邀参与了AAM组织的“ANDA及补充申请常见缺陷”小组讨论。讨论涉及ANDA提交相关问题和审评问题,重点关注质量相关缺陷 。
FDA立卷审查部(Division of Filing Review)主任Johnny Young指出,目前约4%(已从往年的20%大幅改善)的ANDA 在提交时被判定为不完整,导致发出拒收函(Refuse-to-Receive ,RTR)。其中,约21%的ANDA因稳定性 数据不完整被拒绝,19%因未通过Q1/Q2评估被拒绝,16%因356h表格(该申请表格描述提交的原因和主要内容用于录入FDA系统)不完整或不正确被拒绝。
Pollock指出,FDA法规21 CFR 314.101(d) 规定,FDA可以拒绝接收未填写完整申请表的ANDA。尽管法规中“可以”(may)一词的解释存在争议(并非指企业可自决,但至少FDA可行使裁量权),但FDA将其解释为“确定”(definitive),即如果356h表格不完整或不正确,FDA将拒绝受理ANDA。Pollock认为鉴于当前技术条件(即FDA可以通过信息工具与ANDA申办者 保持密切沟通),FDA完全可以要求申办者迅速修正356h表格,而不是直接拒收 导致申办者损失25%的GDUFA 申请费,按2026财年ANDA 申请费358,247美元的25%计算,即89,562美元。
停摆影响检查安排,最常见缺陷还是源自偏差 和OOS
会上,FDA检查与调查办公室(OII,由ORA等检查部门改组而来 )副主任Elizabeth Miller介绍了政府停摆对OII核心活动的影响,包括检查、调查和进口监管。
由于拨款中断,OII需基于风险优先安排活动,包括保障生命安全的检查、有因检查、批准前检查 、GDUFA资助的监管检查、其他用户费用资助的检查以及其他必要的调查活动。她还讨论了OII最近的组织结构调整,该调整旨在通过提供不同层次的检查人员职业路径、培训平台现代化、扩大技术利用以及为检查员提供持续学习机会,实现更灵活的劳动力部署,即识林此前报道的FDA将回归“通才”检查员培养模式 。
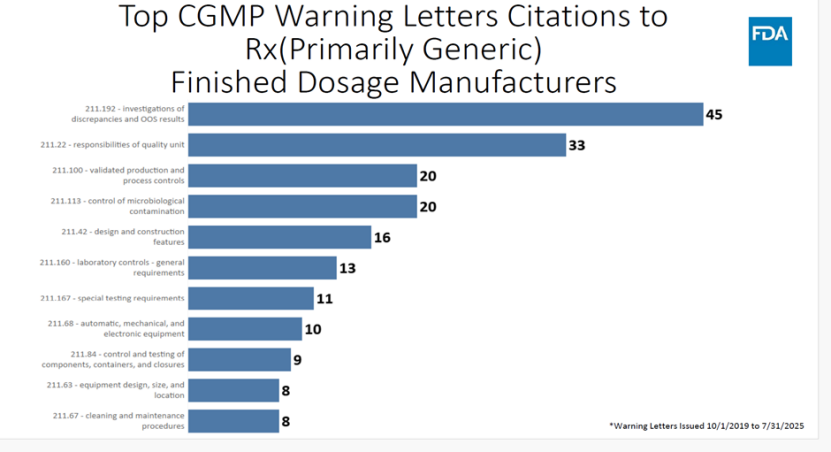
会上也分享了仿制药 公司收到的CGMP 警告信分析,常见缺陷项(对应美国cGMP条款 引用)如下图:
调查差异(discrepancies)和超标(OOS )(211.192):45次
还有生物类似药的里程碑
生物类似药 本是会议两大主题之一,但Pollock仅提到会议期间发布的一份指南引发广泛讨论,可见其影响力之大。该指南就是识林周一报道的FDA发指南建议免去生物类似药疗效比对研究 。在此不再赘述,读者可点击跳转阅读。
识林-实木
识林® 版权所有,未经许可不得转载
适用岗位必读指南:
QA:负责确保所有操作符合cGMP要求,包括生产、质量控制、设备维护等。 生产:必须遵守书面程序,确保产品质量。 质量控制(QC):负责样品的测试和批准或拒绝,以及稳定性测试。 设备维护:确保设备清洁、维护和校准符合规定。 仓储与分销:遵守药品存储和分发的书面程序。 文件适用范围:
文件要点总结:
质量控制单元的责任: 必须有一个质量控制单元,负责批准或拒绝所有组件、药品容器、包装材料、标签和药品,并审查生产记录以确保没有错误发生或错误已得到全面调查。
人员资质与责任: 参与药品生产、加工、包装或储存的人员必须具备相应的教育、培训和经验,并遵守良好的卫生习惯。
设备设计、清洁与维护: 设备应适当设计,便于操作、清洁和维护,并按规定进行定期清洁和维护。
组件和药品容器的控制: 必须有书面程序详细描述组件、药品容器和闭合件的接收、识别、存储、取样、测试和批准或拒绝。
生产和过程控制: 必须有书面程序确保药品具有其声称或代表的身份、强度、质量和纯度,包括偏差处理和产量计算。
包装与标签控制: 必须有书面程序确保正确的标签和包装材料用于药品,包括防篡改包装要求。
仓储与分销程序: 必须有书面程序描述药品的存储和分发,确保药品质量。
实验室控制: 必须建立科学合理的规格、标准、抽样计划和测试程序,以确保药品及其组件符合适当的身份、强度、质量和纯度标准。
记录与报告: 所有与生产、控制或分发相关的记录必须保存至少一年,或在特定情况下保存更长时间,并随时可供授权检查。
退回和报废药品的处理: 退回的药品必须被识别并保留,除非证明其符合适当的安全、身份、强度、质量和纯度标准,否则应销毁。
以上仅为部分要点,请阅读原文,深入理解监管要求。