首页
>
资讯
>
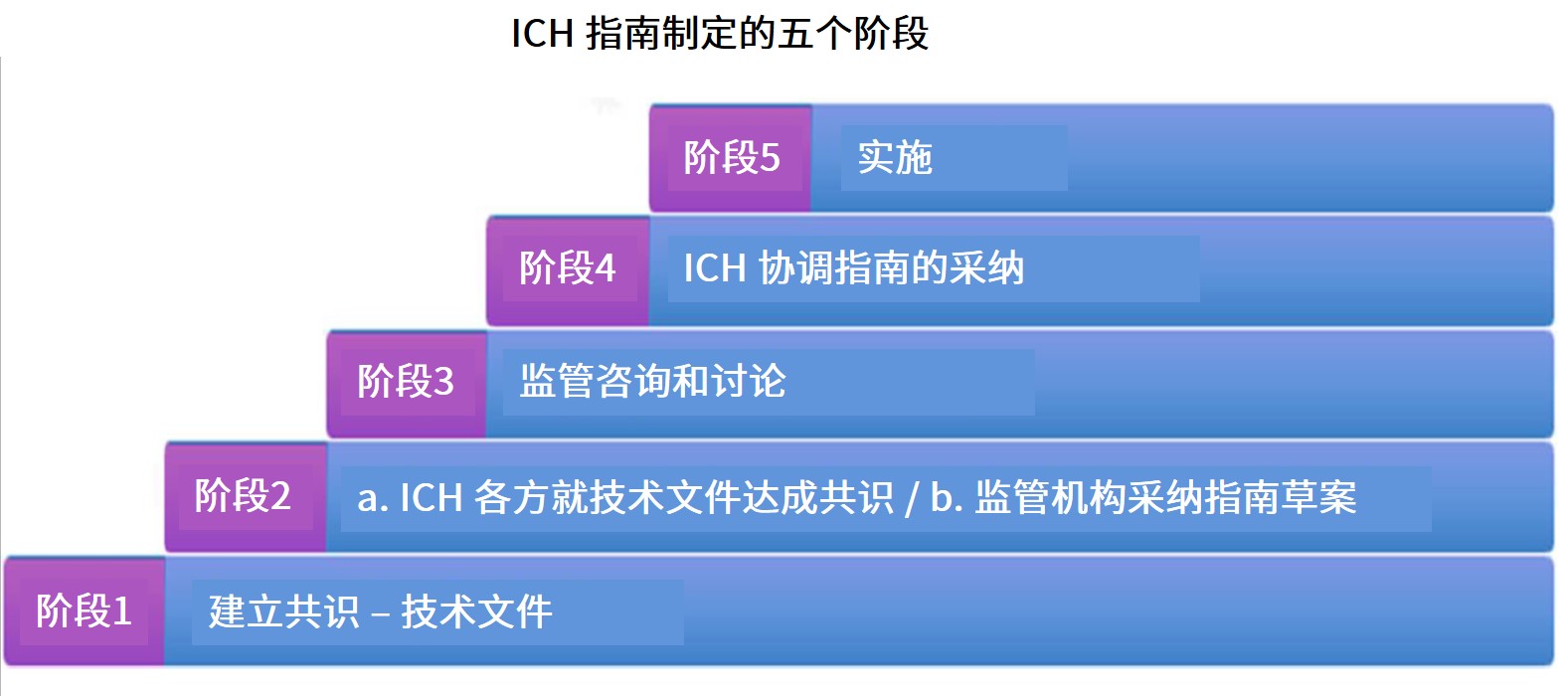
ICH 将于近期发布和起草的指南一览
出自识林
2019-05-28
美国 FDA 于 4 月 29 日上午与加拿大卫生部联合举办了 ICH 公开咨询会议,简要介绍了十几份不同 ICH 指南的进展和可能的预期。
在这个持续三小时的会议上,三位 FDA 专家和北卡罗来纳大学生物统计学教授 Lisa LaVange 讨论了近期进入 ICH 流程第 3 阶段的指南主题。FDA 战略项目办公室主任提供了关于电子化标准方面的更新。会议的最后,FDA 中心主任办公室运营研究分析师 Amanda Roache 快速浏览了 ICH 正在开展的工作和议题。
E8(R1) 临床试验的一般考虑
LaVange 讨论了 E8(R1),这是涉及临床试验的一般性考虑以及更广泛的临床质量管理规范(GCP)改革的一部分的修订指南,其中包括采用质量源于设计 框架开展临床研究 ,并扩展指南的覆盖范围,以包括更广泛的研究设计和数据来源。
她表示,专家工作组(EWG)曾三次面对面开会,并举行多次电话会议,希望确保指南紧跟当今世界趋势,现在有各种临床试验设计并且能够使用更多的真实世界数据来源(包括电子健康记录)。她还表示,EWG 曾开玩笑要把该指南重新命名为 E0,“因为这份指南应该先于所有其它指南。”
该指南已于 5 月 9 日进入阶段 3。FDA 将于今年 11 月召开关于该主题的公开会议,并计划于 2020 年 6 月完成第 4 阶段文件。
E19 安全性数据收集的优化
FDA 药品审评第 II 办公室主管 Mary Thanh Hai 讨论了有关处理安全性数据收集的优化问题的 E19。更具体地说,这份 10 页的指南涉及在一些上市前后期或上市后研究中何时使用选择性安全数据收集方法是适当的
尽管 E19 不会改变地区/区域安全性报告要求,但申办人 和研究人员仍应确保常规患者护理不会受到影响。Hai 解释了如何在试验的某些患者亚组中进行选择性安全性数据收集,或者当一个药已经被一个监管机构批准,且公司希望向新监管机构提交相同适应症 时进行选择性安全性数据收集。她还强调,应尽早与监管机构协商,采取这种选择性方法。
目前,E19 正处于第 3 阶段,欧盟将在 9 月 29 日之前提交反馈意见。Hai 表示,EWG 计划明年秋季召开会议审查公众意见,,并于 2021 年 6 月进入第 4 阶段。
S11 支持儿科药物开发的非临床安全性测试
FDA 新药办公室的 Karen Davis Bruno 介绍了 S11 的进展,该指南涉及儿科药物的非临床安全性测试,目前正处于 ICH 流程的第 3 阶段。
她表示,该指南将于 6 月份在阿姆斯特丹举行的面对面会议上讨论,以审查于 4 月初截止提交的公众意见。行业组织最近呼吁修订 S11 ,减少对幼年动物研究的需求。
M10 生物分析方法验证
FDA 临床药理学处副处长 Brian Booth 讨论了 M10 生物分析方法验证。这份长达 57 页的指南自 2016 年底开始制定,为化学和生物药定量的生物分析 测定验证以及其在非临床和临床研究的研究样本分析中的应用提供建议。
Booth 表示,他希望专家们能够在 11 月就指南开展面对面讨论,并在 2020 年 11 月之前使指南进入第 3 和 第 4 阶段。除日本药品与医疗器械管理局(PMDA)(正在翻译指南)和 FDA 尚未开始征询意见外,所有其它主要监管机构已开始就 M10 征询意见。
电子标准主题和 MedDRA 更新
FDA 战略项目办公室主任 Mary Ann Slack 提供了关于 E2B(R3) 临床安全性资料的管理:个案安全报告传输的数据要素 ,M2 监管信息传输的电子标准 ,以及 M1 监管工作医药词典 (MedDRA )。
关于 E2B(R3),Slack 表示,他们正在编写培训材料并准备在 E2B 和欧洲药品质量管理局(EDQM)术语之间绘制管理路线。她指出,他们还计划与 FDA 正在开发的 FAERS II 合作,并将在 2020 年上半年与行业测试推出 E2B(R3)。
M2 工作组与评估、谷歌以及其他科技公司一起,正在制定关于 HL7 快速医疗可互操作性资源的白皮书,并且正在开展相关 ICH 考量的工作。她还指出,M2 的主管范围包括监测影响 ICH 关注领域的技术和监管趋势,例如管理与标准制定组织的关系,包括 HL7、ISO/TC215 和 EDQM。
就 MedDRA 而言,她表示,现在已经有 110 个国家的 5000 多个组织机构订阅了 MedDRA,并指出,预计将在包括中美洲、韩国、中国和印度在内的几个新区域进一步提供本地化支持。此外,俄语 MedDRA 翻译已经完成,韩语翻译正在进行中。重点关注数据质量和用药错误的 MedDRA 考量要点配套文件拟定于今年更新。
更多指南
FDA 中心主任办公室运营研究分析师 Amanda Roache 还谈到了今年或不久的将来即将推出的一系列指南,但她指出,时间表可能会有所变化。
M12 药物相互作用研究
Roache 表示,该指南主要协调用于设计、实施和开展药物相互作用 (DDI)研究的方法,DDI 研究旨在评估治疗产品开发过程中 DDI 的潜在可能。指南还将在评估体外和体内 DDI 研究方面协调监管期望。M12 预计将于今年 6 月起草推出。
E20 自适应临床试验
E20 主要是协调有关自适应临床试验涉及的规划、实施和监管审评的监管视角,为自适应试验设计定义了一套指导原则,指导设计、实施、分析和解释的所有方面。 该指南同样将于今年 6 月推出。
E9(R1) 附录:临床试验统计原则
Roache 表示,对于这一附录,工作组正在寻求建立框架工作,将试验目标转化为对估计治疗效果的精确定义。除了澄清现有的 E9 外,监管机构还将对其扩展并重新审视缺失数据和分析的问题。最终指南将于今年年底完成。
E11A 儿科外推
该指南旨在降低初始成人批准与在产品标签中纳入儿科特定信息之间的巨大差距(7-10 年)。 此外,指南还将协调把儿科外推纳入整体药物开发计划的方法和策略。指南草案预计将于 2020 年 11 月完成。
E14/S7B 问答:非抗心律失常药物致 QT-QTc间期延长及潜在致心率失常作用的临床和非临床评估
该问答指南旨在简化那些延长 QT 间期但致心率失常风险较低药品的临床开发,并且可能帮助在开发中放弃更少的候选产品。 该文件于 2018 年起草,预计将于 2020 年 6 月完成第 1 阶段定稿。
M9 基于生物药剂学分类系统的生物等效性试验豁免
预计将于今年完成定稿指南(指南草案于 2018 年发布)。她表示,指南将着眼于提供药品的生物药剂学分类 ,并在当体内研究对于证明药品生物药剂学质量是不必要的时候,减少药物开发的成本和时间。
M7(R2) 附录:评估和控制药物中的 DNA 反应性(致突变)杂质以限制潜在的致癌风险
Roache 表示,修订该指南的专家正在开展两个不同的项目:附录和问答文件。问答文件预计将于今年发布。开发问答文件是为了澄清和解决 2014 年 M7 实施后发现的质量和安全问题以及相关关注点。
M11 协同的电子结构化临床试验方案(CeSHarP)
S1(R1) 人用药的啮齿动物致癌性研究修订
于 2013 年 8 月开始的一项研究的结果将决定是否需要修订该指南。Roache 表示,需要确定的是,是否可以使用证据权重方法来表征致癌性风险,而不开展为期两年的大鼠致癌性研究 。
S5(R3) 关于人用药生殖毒性检测的修订
该指南正在根据收到的意见进行修订,预计将于 11 月发布定稿指南。
Q3C(R8) 残留溶剂指南 维护
该指南将制定三种溶剂的 PDE 水平:2-甲基四氢呋喃,环戊基甲基醚和叔丁醇。指南草案预计将于 2019 年底发布。
Q3D(R2) 元素杂质指南 维护
正在开展的工作包括用于皮下和透皮给药路径 的 PDE。指南草案将于今年下半年发布。
Q12 药品生命周期管理的技术和监管考虑
指南将提供框架指南,以便在整个产品生命周期 内以更可预测和更有效的方式管理批准后 CMC 变更 。指南还将允许监管机构能够更好地了解公司批准后 CMC 变更管理的药品质量体系 。指南草案于 2017 年 11 月发布,目标是在今年 11 月之前完成定稿。
Q13 连续制造
作为减少实施连续制造 障碍的努力的一部分,该指南主题于 2018 年 6 月启动 , 预计将于 2020 年 6 月发布指南草案。
非正式讨论组
另外,2019 年,ICH 还建立了两个新的非正式讨论组 , 一个是关于质量方面,查看与质量相关的问题以协调产品生命周期内的质量管理体系;另一个是关于仿制药 , 考虑需协调的仿制药相关指南的具体领域和可能性,例如生物等效性指南。【ICH 正式发文计划协调仿制药标准 2019/02/08】
作者:识林-椒® 版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
参考资料 Health Canada and United States Food and Drug Administration Joint Public Consultation on ICH. FDA. 会议幻灯片:Overview of Ongoing ICH Topics 20190429 ICH Updates: What’s Coming in 2019 and Beyond. ICH.
适用岗位:
研发(R&D) :应密切关注ICH M7指南中对DNA反应性(突变性)杂质的评估和控制要求,确保在药物研发过程中识别、分类、鉴定和控制这些杂质,以限制潜在的致癌风险。注册(Reg) :需理解并应用ICH M7指南,为新药物质和新药产品在临床开发和市场申请中提供合规性支持,特别是在药物合成、配方、组成或制造过程发生变化时。质量管理(QA) :必须遵守ICH M7指南中对突变性杂质的控制策略,确保产品质量和安全风险管理。工作建议:
研发(R&D) :在药物合成和制剂开发过程中,主动识别和评估可能产生的突变性杂质,制定相应的控制措施,并在临床前研究中考虑这些杂质的潜在影响。注册(Reg) :在准备临床试验申请和市场申请时,确保包含有关突变性杂质的评估和控制策略,以满足监管要求。质量管理(QA) :建立和维护一个控制策略,包括对原材料、中间体和最终产品的检测,以及对生产过程的监控,以确保突变性杂质控制在可接受的水平。适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
生物分析科学家(Bioanalytical Scientist):深入理解生物分析方法验证的各个环节,确保实验设计和操作符合指南要求。 质量保证专员(QA):监督生物分析方法验证流程,确保合规性。 研究与开发人员(R&D):在药物开发过程中应用验证的生物分析方法,确保数据的可靠性和一致性。 注册专员(Regulatory Affairs Specialist):了解验证要求,为药品注册申报准备相应的文件和数据。 文件适用范围:
要点总结:
方法验证目的 :明确生物分析方法验证旨在证实方法适用于其预定目的,强调方法的适用性和准确性。选择性和特异性 :强调了生物分析方法在存在潜在干扰物质时区分和测量药物的能力,确保结果的可靠性。校准曲线和范围 :详细规定了校准曲线的构建和验证,包括对最低定量限(LLOQ)到最高定量限(ULOQ)的全面评估。准确度和精密度 :通过质量控制样品(QCs)的评估,确保分析结果的准确度和精密度符合预定标准。稳定性和重现性 :对样品在不同条件下的稳定性进行了规定,包括冻融稳定性、短期稳定性和长期稳定性,以及重复注射的重现性。以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA:负责确保质量管理体系的实施和监督,建议定期审查和更新质量管理体系文件。 生产:确保生产过程符合质量管理体系要求,建议参与设备和工艺管理的持续改进。 研发:在产品设计和开发阶段考虑质量管理体系要求,建议与QA紧密合作以确保合规性。 适用范围:
文件要点总结:
质量管理体系概述 :明确了质量管理体系的发展、基本概念及其相互关系,强调了高层管理者在质量方针、目标和计划制定中的关键作用。产品质量实现要素 :涵盖了机构与人员、厂房设施、设备、物料与产品、工艺管理等关键要素,特别指出了人员培训和设备生命周期管理的重要性。质量保证要素 :包括变更管理、偏差管理、产品质量回顾、投诉和召回管理,强调了CAPA系统在持续改进中的作用。质量风险管理 :介绍了质量风险管理的职责、模式图、流程和步骤,以及在企业和管理机构中的应用。质量管理系统文件 :规定了文件体系结构、生命周期和种类,强调了文件管理在确保质量管理体系有效运行中的重要性。以上仅为部分要点,请阅读原文,深入理解监管要求。