|
首页
>
资讯
>
【识林翻译】PICS检查清单,AQbD系列,FDA早期GMP指南,14份警告信,等
出自识林
【识林翻译】PICS检查清单,AQbD系列,FDA早期GMP指南,14份警告信,等
2022-07-23
| 识林里,中外法规指南浩如烟海。识林用户的第一步,往往是从本职工作出发,检索、阅读和使用。即使没有出口业务的用户,也可开展系统性学习,遍读各国法规,形成对重要知识点的深度认知。为帮助用户跨越语言关,加深理解,识林翻译团队(也包括向导)将努力为识林企业用户提供法规指南翻译,并尽我们所能确保质量。
专业水平所限,差错难免,也希望企业用户在阅读学习的同时,在页面评论区提问、纠偏,还可联系我们提出法规翻译需求。
|
PIC/S系列指南
包括:
PIC/S PI 023–2备忘录 药品质量控制实验室的检查
PIC/S PI 030-1原料药检查 备忘录
PIC/S PI 043-1检查员备忘录 共线生产设备的交叉污染
PIC/S公共设施检查 备忘录
PIC/S与包装相关的GMP检查 备忘录
所谓“备忘录”,可理解为检查员开展工作时所用的Checklist,对企业日常实务具有相当参考价值。识林将继续翻译PIC/S各类指南。中国已开启加入PIC/S进程,企业用户可阅读学习,早做准备。
AQbD系列指南
包括:
MHRA“分析质量源于设计”项目技术回顾
BP英国药典增补章节(征求意见稿):分析质量源于设计(AQbD)应用于药典方法
指南导读:USP<1220>分析方法的生命周期
ICH最近发布的Q14,正式将AQbD(分析质量源于设计)的理念推而广之。但其实,英国MHRA,BP,以及USP已经开始这方面的监管探索。此次翻译的三文,可系统了解AQbD理念的起源。
此外,AQbD和方法学开发的系列课程均已上线。
【ACD - 识林】AQbD系列课程 2022.07
{{#video:v09315531-b956-4e54-9bde-8174816afaba}}
FDA早期GMP相关指南
包括:
FDA指南定稿 制药用水
FDA清洁工艺验证(7/93)
FDA关于注射剂冻干的检查指南
上述指南均发布于90年代,尽管年代看似久远,但如结合当前最新要求做对比解读,可对相关概念和监管方法有更深理解。
FDA警告信
包括:
警告信 马来西亚Furley Bioextracts SDN BHD
警告信 澳大利亚Natural Beauty Care Pty Ltd
警告信 美国Glenn Burkett Naples Corporation
警告信 洪都拉斯Laboratorio Pharma International SRL
警告信 墨西哥Nanomateriales Químicos Avanzados, S.A. de C.V.
警告信 印度Aurobindo Pharmaceutical Limited
警告信 美国Professional Disposables International, Inc.
警告信 韩国Cosmo Bio Co., Ltd.
警告信 美国Crystal Clear Supplements
警告信 美国Richard J. Obiso, PhD dba Avila Herbals, LLC
警告信 美国Vital Health & Wellness
警告信 美国Yusef Manufacturing Laboratories, LLC
警告信 土耳其Gulsah Uretim Kozmetik Sanayi Anonim Sirketi
警告信 美国Joe Wise Pharmacy, Inc., dba Wise Pharmacy
识林近期加快警告信翻译,供质量人员阅读参考。
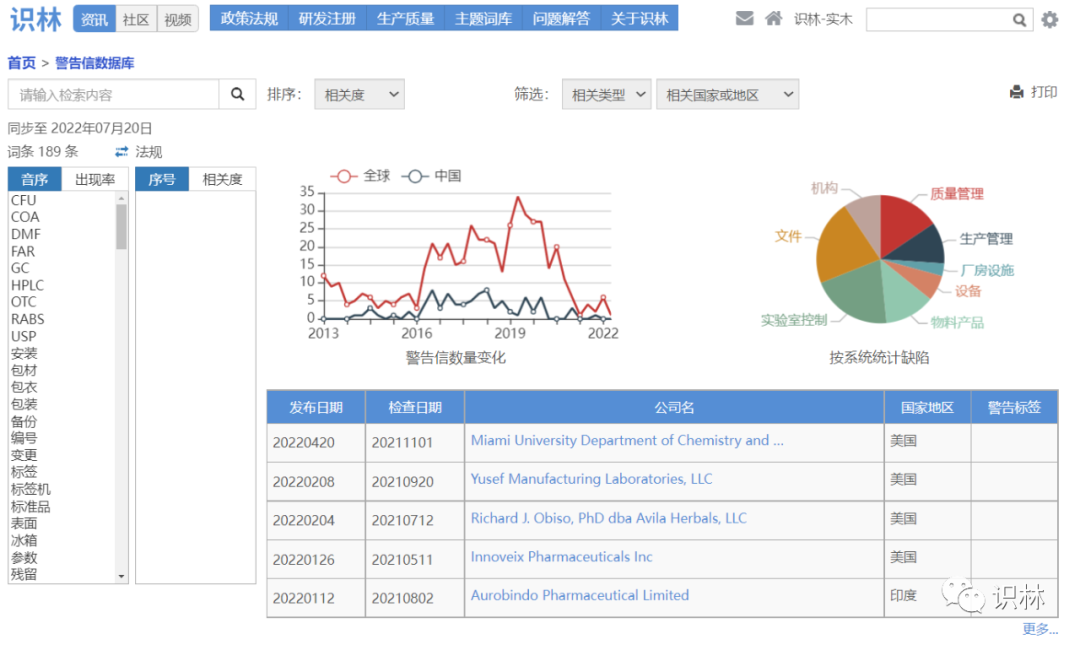
近期警告信可在“最近更新”的“警告信”栏目浏览:
更多警告信案例,可查询“警告信数据库”:
另外,识林已上线“机翻”功能,近期更新的英文法规指南都将提供双语机翻版,帮助用户阅读学习。
识林®版权所有,未经许可不得转载
岗位必读建议: - QA:确保水处理系统和水质符合USP和CGMP要求。
- 生产:监控水处理过程,确保水质适用于药品生产。
- 研发:在药品开发过程中,考虑水质对药品质量的影响。
文件适用范围:
本文适用于药品生产中使用的水,包括化学药、生物制品等,主要针对美国市场,适用于大型药企、Biotech及CRO等。 文件要点总结: - 水质分类与标准:明确了8种不同类型的制药用水及其USP标准。
- 水源与制备:USP WFI需通过蒸馏或反渗透制备,饮用水通常来自市政系统。
- 污染源控制:强调了管道系统可能的污染问题,以及用点取样的重要性。
- 水处理系统要求:详细说明了水处理系统的验证、储罐管理及过滤和消毒措施。
- 水质监测与标准:CGMP要求建立质量标准,监测频率应覆盖季节性变化。
以上仅为部分要点,请阅读原文,深入理解监管要求。 解读法规指南:《注射剂冻干检查指南》 适用岗位: - 生产操作人员(必读):确保冻干过程符合指南要求,注意无菌操作和设备使用。
- 质量保证专员(QA)(必读):监控冻干产品质量,确保生产过程和产品质量符合法规要求。
- 设备工程师:负责冻干设备的维护和验证,保证设备正常运行。
- 研发人员:在产品开发阶段考虑冻干过程的特殊要求。
工作建议: - 生产操作人员:遵循冻干操作规程,注意无菌技术和操作细节。
- QA专员:制定检查计划,确保产品在生产、检验各环节符合标准。
适用范围:
本文适用于通过冻干技术生产的无菌注射剂,包括抗生素、生物技术产品等。主要针对美国FDA监管下的药品生产企业,特别是Biotech和大型药企。 要点总结: - 冻干过程控制:强调了冻干过程包括冷冻、一次干燥(升华)和二次干燥(解吸)三个阶段,对产品质量至关重要。
- 无菌操作:明确了在冻干过程中,无菌操作是防止产品污染的关键环节。
- 设备验证:规定了冻干设备必须经过验证,包括冻干周期的验证和设备无菌保证。
- 产品稳定性:指出了冻干产品在稳定性方面的考虑,包括水分含量和剂量均匀性测试。
- 最终产品检查:强调了对冻干产品的最终检查,特别是对融解回现象(meltback)的关注,因为这可能影响产品的稳定性和效力。
以上仅为部分要点,请阅读原文,深入理解监管要求。 必读岗位及工作建议- QA(质量保证):应详细阅读并理解清洁工艺验证的指南,确保企业内部的清洁流程与FDA的要求一致。
- 生产操作人员:必须熟悉清洁工艺的SOP,按照验证过的程序执行清洁工作。
- 验证工程师:负责制定和执行清洁工艺的验证方案,确保验证结果的准确性和可靠性。
- 研发:在开发新药或改变生产工艺时,考虑清洁工艺的验证需求,确保新产品的顺利上市。
文件适用范围本文适用于原料药和成品制剂行业的设备清洁验证,特别关注化学残留物的清洁。适用于化学药品、生物制品等药品类型,包括创新药和仿制药。发布机构为美国FDA,适用于Biotech、大型药企、跨国药企等企业类别。 文件要点总结- 清洁工艺验证的必要性:强调了清洁工艺必须经过验证,以确保清洁效果符合预期,防止药品污染或掺杂。
- 验证过程的目标和方法多样性:指出清洁验证的目标是确保系统一致性,并认可存在多种验证方法。
- 书面规程和验证方案:规定企业必须有详细的书面清洁工艺和验证方案,包括责任分配、接受标准和再验证要求。
- 限度的建立:强调企业需基于科学依据建立合理的残留物限度,FDA不设定具体标准。
- 清洁验证的评估:包括设备设计、清洁工艺的书面记录、分析方法的专属性和灵敏度、取样方法等,以确保清洁工艺的有效性。
以上仅为部分要点,请阅读原文,深入理解监管要求。 法规指南解读 一、适用岗位及工作建议: - 本文件为PIC/S发布的《共线设施的交叉污染检查员备忘录》,适用于以下岗位:
- QA(质量保证部门):必读。应确保所有GMP检查包含交叉污染控制的QRM,并与产品的危害等级相适应。
- 生产部门:必读。应根据产品的危害等级和生产设施的特点,制定和执行适当的组织和技术措施。
- 研发部门:参考。在新产品设计和开发阶段,考虑共线生产可能带来的交叉污染风险。
- 注册部门:参考。在药品注册过程中,需考虑和说明共线生产对药品安全性、质量和效力的影响。
二、适用范围: - 本文适用于化学药品、生物制品、原料药等的生产,特别针对在共用设施中生产具有“较高危害”等级的起始物料的产品。适用于不同PIC/S成员国的制药企业,包括Biotech、大型药企、跨国药企等。
三、要点总结: - 风险管理的重要性:“应当”识别、设计并实施基于危害的风险控制措施,通过QA系统结合GMP进行有效监控和定期审查。
- 检查员的评估重点:在GMP检查中,检查员“应评估”交叉污染风险管理的系统性过程,包括危害评估的完整性和设施设计的适宜性。
- 技术和组织措施:强调了技术措施(如厂房和设备设计)和组织措施(如阶段性生产和清洁确认)的重要性,以及它们在减少交叉污染风险中的作用。
- 创新与灵活性:提到了制药生产设备和技术的进步,为共线生产带来更大的灵活性,但“必须”确保操作在必要时完全和全面地隔离。
- 健康基础限值:提及了为患者或目标动物设置基于健康的允许暴露限值,但“不规定”具体方法,相关指南在PIC/S PI 046中有详细说明。
以上仅为部分要点,请阅读原文,深入理解监管要求。 岗位必读建议: - QA(质量保证):确保理解包装相关GMP检查的关键方面,以及如何使用质量风险管理工具进行合规性评估。
- 包装操作人员:了解包装过程中的GMP要求,包括设备操作、清洁、物料平衡和偏差调查。
- 质量控制(QC):掌握包装材料的质量标准和测试方法,以及无菌产品包装完整性的评估。
文件适用范围:
本文适用于化学药、生物制品等药品的包装和标签过程的GMP检查,包括无菌产品和非无菌产品。适用于各种企业类别,包括Biotech、大型药企、跨国药企等,由PIC/S发布,反映了2005年至今的技术水平。 文件要点总结: - 供应商管理: 强调了对供应商批准、审计和物料规格符合性的重要性,以及供应商交货过程中的质量保证措施。
- 接收与存储: 明确了接收时的质量文件检查、包装完整性和鉴别,以及存储区域的组织、清洁状态和物理隔离。
- 质量控制: 规定了包装材料的采样计划、实验室设备、功能测试和完整性测试,以及与内包材质量相关的中间过程控制。
- 生产场所与设备: 强调了包装区域的设计、环境监控、公用设施确认和包装设备的性能、维护与验证。
- 文件与记录: 规定了批包装记录、物料平衡SOP、行动限制和包装过程偏差的调查,以及电子数据输入的安全性。
以上仅为部分要点,请阅读原文,深入理解监管要求。 岗位必读建议: - GMP检查员:必读。在培训和检查准备中使用本文件,以优化检查时间和评估GMP合规性。
- 设施管理:必读。确保HVAC、制药用水、蒸汽和医用气体系统符合GMP要求。
- 质量保证(QA):必读。监督和审核相关系统和流程的GMP合规性。
- 生产操作:必读。了解操作区域/项目的具体要求和标准操作程序(SOP)。
工作建议: - GMP检查员:使用本备忘录作为检查的参考,确保检查的全面性和系统性。
- 设施管理:定期评估和优化设施设计和操作,以符合最新的技术和GMP要求。
- 质量保证(QA):基于本文件进行内部审计,确保所有相关领域都达到GMP标准。
- 生产操作:遵循SOP进行日常操作,及时报告任何偏差或不符合GMP要求的情况。
文件适用范围:
本文适用于制药行业中的HVAC系统、制药用水、蒸汽和医用气体的GMP检查,不特定于药品类型或注册分类,由PIC/S发布,适用于追求GMP合规性的企业,包括Biotech、大型药企、跨国药企、CRO和CDMO等。 文件要点总结: - HVAC系统设计关键参数:强调了独立系统需求、过滤等级、气流方向等设计要素。
- 系统确认:包括DQ、IQ、OQ、PQ和必要时的计算机验证,确保系统性能符合要求。
- 现场巡视:检查设计规范、图纸与实际的差异,以及计划外维护和变更控制。
- 监测与质量控制:涵盖了环境监测、化学残留检测、质量控制检验和趋势分析。
- 维护、校准和文件记录:强调了维护计划、校准计划、SOP、记录和再确认的重要性。
以上仅为部分要点,请阅读原文,深入理解监管要求。 法规指南解读 适用岗位: 工作建议: - QA:确保原料药检查流程符合GMP要求,监督实施。
- 生产:按照检查要素准备和执行原料药生产。
- 研发:在原料药开发阶段考虑检查要求,确保可检查性。
- 注册:了解监管要求,确保注册文件符合检查标准。
文件适用范围:
本文适用于原料药(APIs)的生产检查,包括化学药、生物制品等,特别关注多功能设施和最终生产步骤的污染控制。适用于遵循PIC/S指导的成员国,包括Biotech、大型药企、跨国药企等。 要点总结: 原料药检查准备:强调检查前对生产场地、产品、流程和设备的详细了解,包括API的分类、预期用途、生产批量等。 质量管理体系:明确内部审计、变更控制、偏差处理等关键环节的管理要求,确保质量体系的有效运行。 厂房和设施:对原料药生产环境的控制,包括储罐区、清洁室、控制室等特定区域的管理,以及防止污染和交叉污染的措施。 工艺设备:虽然本部分内容将在下一版修订中制定,但强调了对工艺设备的检查和维护的重要性。 文件和记录:要求对生产过程中的关键文件和记录进行严格管理,确保数据完整性和可追溯性。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |