|
首页
>
资讯
>
识林年度回顾-2018 FDA 药品指南制定计划和完成
出自识林
识林年度回顾-2018 FDA 药品指南制定计划和完成
2018-12-29
2018年马上就要过去了,12月22日开始,FDA因处于资金中断的特殊时期,只会对一些紧急的公共卫生安全威胁等法律允许的范围内继续运行,又正处于圣诞节和元旦的假期之中,剩下的3天基本不会有新指南发布,CDER年初发布的计划完成的怎么样呢?识林带大家一起来回顾一下。
2018年计划与完成
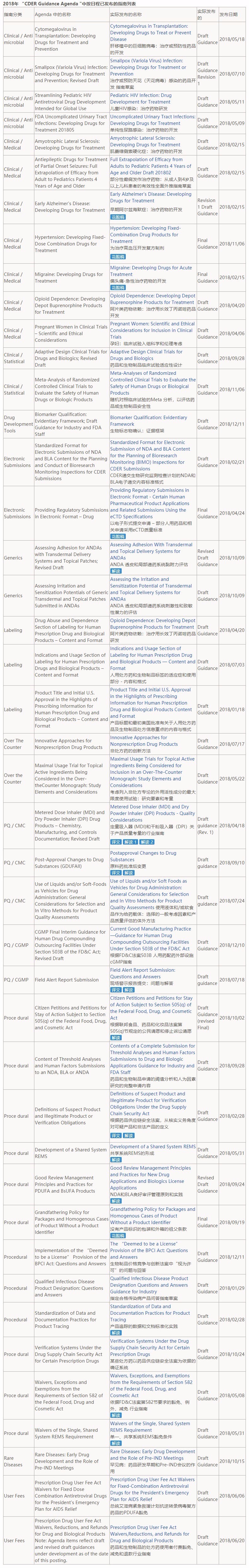
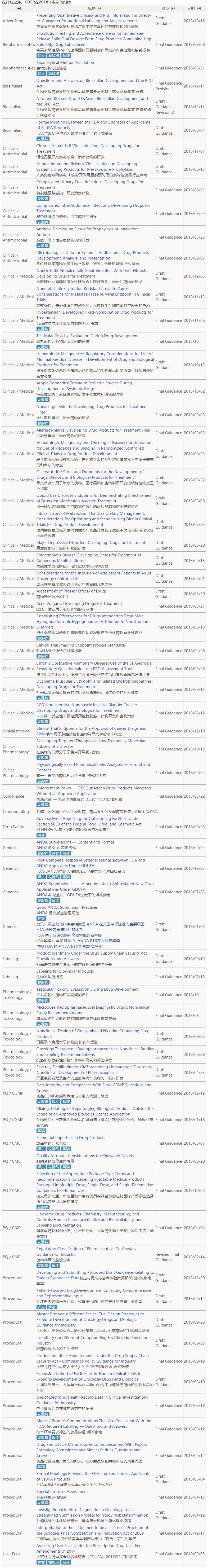
CDER在2018年1月16号发布的2018年度指南计划中列出了98篇指南,程序类指南有24篇占比重为25%,药品质量/CMC、仿制药类别指南分别是13篇和10篇,药品质量/生产标准(CGMP)的计划是2篇。
截至2018年12月28日,FDA完成了计划中的43篇,占计划的44%,而且FDA还发布了71篇不在2018年度计划内的指南,其中有些是在完成2017年度或更早前的计划(43+71篇目详情请见文末列表)。
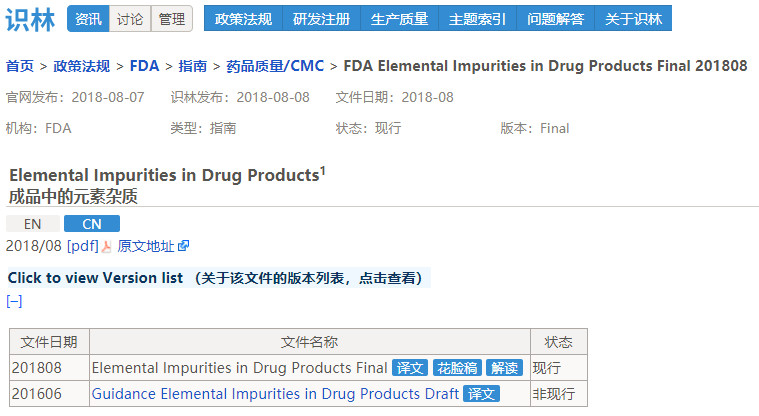
药品质量/CMC 指南亮点:元素杂质、原料药变更和特殊产品
在CMC分类中的指南,计划发布13篇,这当中令人格外关注的指南有以下篇目1-6, 2018年发布了其中的3、4、5条,2和6自2016年就开始出现在每一年的计划中。计划外发布的篇目有7-11条。
即全年发布CMC指南8篇,识林中为每一篇指南都做了中译或花脸稿、解读等工作,以《成品中的元素杂质》指南为例,通过版本列表可以了解该篇指南的 “前世今生”,花脸稿用红色、删除线标记了新旧版本的变化之处,并且配以导读进行说明,例如草案中“如果一种元素杂质在成品中含量超过PDE的可能性足够小,那么这种杂质在日常检测中就不需要检测”,在此次的定稿中的表述改为“可能没有必要检测每批药物的杂质”。
仿制药指南:关键指南基本迟到,复杂仿制药独受关注
2018年的计划中仿制药分类指南有10篇,篇篇受人瞩目,其中只发布了两篇有关透皮和外用递药系统(TDS)仿制药的指南。
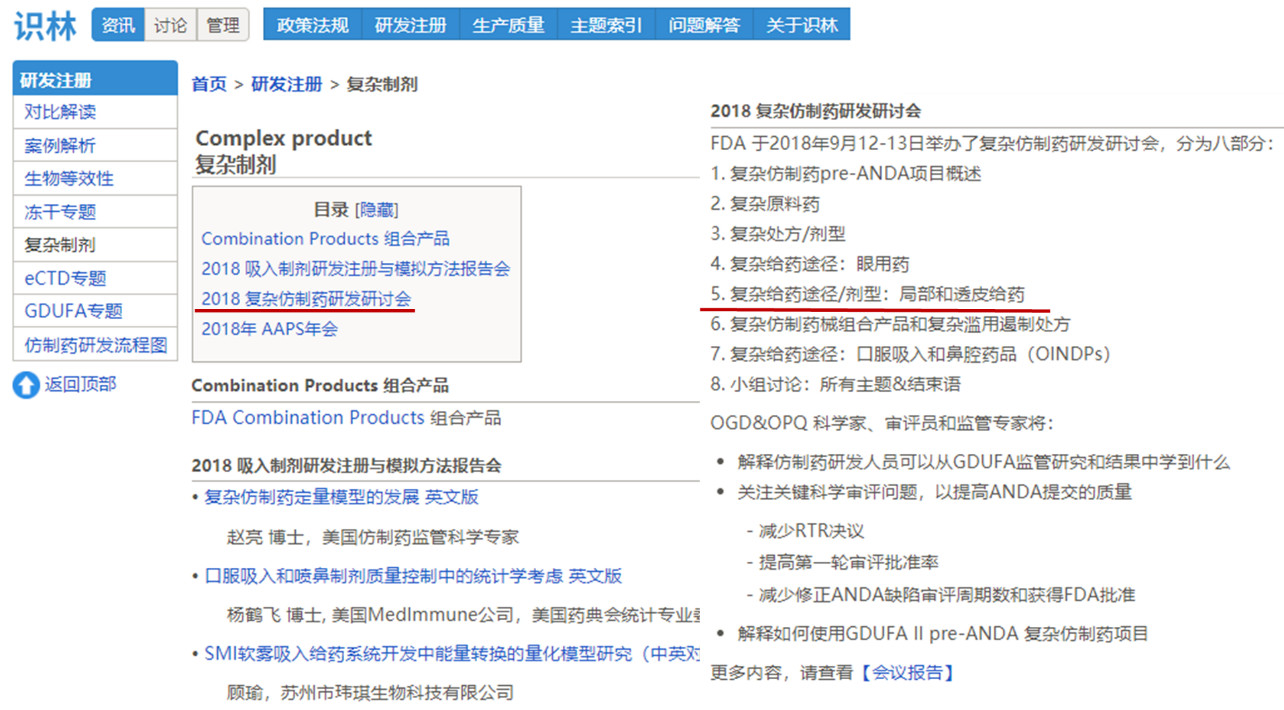
尽管仿制药指南发的不尽如人意,但FDA 2018年对复杂仿制药的关注可见一斑,除了上述两篇指南和前文提到的吸入式制剂CMC指南,FDA举办了为期2天的《2018 复杂仿制药研发研讨会》。
因此,识林新建立的复杂制剂专题中,除上述内容外,还收录了北京大学《2018吸入制剂研发注册与模拟方法报告会》,以及美国药学科学家协会中国讨论组和国际药用气雾剂联盟主办的《国际药物质量研究前沿技术论坛》的会议报告内容。
药品质量/生产标准 (CGMP)
也许是任务不多,也许是表现稳健,CGMP方面共发布4篇指南,不仅全额完成了任务,还加倍超额完成。
值得一提的是,《现场警示报告提交》回答了许多令人头疼的问题,美国仿制药界资深人士 Bob Pollock 先生在其博客中表示,“在我 44 年的职业生涯中,我认为没有任何问题让企业抓耳挠腮的焦虑程度能够超出 FAR,现在 FDA 终于发布了一份问答指南。”
此外,《药品CGMP数据可靠性与合规的问题与解答》定稿也备受关注,与草案相比变化很大。更多详情请登录识林参考指南中译、花脸稿及其解读。
附:43+71篇指南列表
作者:识林-枫 识林-樟 识林-榕
岗位必读建议: - QA:需深入理解数据完整性在CGMP中的重要性,并确保所有数据的可靠性和准确性。
- 生产:应了解CGMP工作流程的验证要求,以及如何通过验证确保生产记录的准确性。
- 研发:需掌握数据完整性的概念,确保实验数据的完整性和可追溯性。
- 信息技术:应熟悉CGMP计算机系统的访问限制和数据安全要求。
文件适用范围:
本文适用于包括生物制品在内的药品CGMP,由美国FDA发布,适用于所有在美国市场运营的药企,包括Biotech、大型药企、跨国药企等。 要点总结: - 数据完整性的定义与重要性:强调数据完整性(ALCOA)对于确保药品安全、效力和质量的重要性。
- CGMP记录的术语解释:明确了数据完整性、元数据、审计追踪等关键术语在CGMP记录中的具体含义。
- 计算机系统验证:规定了CGMP工作流程在计算机系统中的验证要求,以及如何通过系统设计和管理确保数据的完整性。
- 审计追踪的审查:强调了审计追踪的重要性,以及审查审计追踪的频率和责任人。
- 数据完整性问题的处理:提出了FDA对数据完整性问题的看法,包括调查、风险评估和纠正措施。
以上仅为部分要点,请阅读原文,深入理解监管要求。 岗位必读建议: - QA:应深入理解数据完整性与CGMP合规性的关系,确保所有CGMP记录符合数据可靠性要求。
- 研发:在实验设计和数据记录过程中,需确保数据的原始性、完整性和准确性。
- 生产:在生产过程中,必须遵循CGMP规范,确保所有操作和记录的合规性。
- 质量控制:负责审核审计追踪,确保数据的准确性和完整性。
- 信息技术:需确保计算机系统的数据完整性,包括访问控制和系统验证。
文件适用范围:
本文适用于化学药品和生物制品的CGMP合规性,包括原料药、成品制剂以及PET药物。主要针对美国市场,由FDA发布,适用于Biotech、大型药企、跨国药企等。 文件要点总结: - 数据完整性定义:强调数据的完整性、一致性和准确性,要求数据在整个生命周期中符合ALCOA原则。
- 审计追踪的重要性:明确审计追踪是重建电子记录事件过程的关键,必须安全、有时间戳,并与元数据一起保存。
- 计算机系统验证:指出每个CGMP工作流均需验证,以确保系统符合预期用途,包括软件、硬件、人员和文件记录的控制。
- 数据可靠性问题处理:建议企业通过调查确定问题范围和根本原因,进行风险评估,并实施管理策略。
- 员工培训:强调应将防止和发现数据可靠性问题纳入员工的常规CGMP培训计划。
以上仅为部分要点,请阅读原文,深入理解监管要求。 适用岗位: - QA:必读,需确保生产过程和产品质量符合USP<232>和<233>以及ICH Q3D指南的要求。
- 注册:必读,负责将元素杂质控制措施纳入药品注册文件,并处理与此相关的变更申请。
- 研发:必读,负责进行元素杂质的风险评估,并根据评估结果调整产品配方或生产工艺。
- 生产:必读,需在生产过程中实施元素杂质的控制措施,并确保符合相关标准。
适用范围:
本文适用于在美国上市的化学药品和生物制品,包括创新药、仿制药、生物类似药及原料药。适用于Biotech、大型药企、跨国药企以及CRO和CDMO等企业类别。发布机构为FDA。 要点总结: - ICH Q3D与USP<232>、<233>的一致性:强调了药品生产企业应遵循ICH Q3D指南,同时满足USP<232>和<233>中关于元素杂质控制的要求。
- 风险评估的重要性:明确指出生产企业应进行风险评估,以确定元素杂质的潜在来源,并据此建立控制措施。
- 实施日期与变更要求:规定了2018年1月1日为元素杂质控制的实施日期,并要求对已批准的NDA和ANDA进行相应的变更。
- 分析方法的选择与验证:强调了选择适当的定量分析方法对元素杂质进行检测,并要求对这些方法进行验证。
- 文件记录与现场检查:要求生产企业记录风险评估和控制措施,并在FDA现场检查时提供相关文件。
以上仅为部分要点,请阅读原文,深入理解监管要求。 岗位必读建议: - NDA/ANDA申请人:必须熟悉FAR提交流程和要求,确保在规定时间内提交FAR。
- QA(质量保证):应监督FAR的提交过程,确保符合FDA的监管要求。
- 研发:在药品开发阶段了解FAR要求,确保产品信息准确无误。
- 生产:在生产过程中注意任何可能导致FAR提交的情况,并及时通报。
文件适用范围:
本文适用于通过NDA或ANDA批准的化学药品和生物制品,包括在美国境内外分销的产品。适用于所有NDA和ANDA申请人,包括Biotech、大型药企、跨国药企等。由美国FDA发布。 文件要点总结: - FAR定义与触发条件:FAR作为早期预警系统的一部分,要求在特定问题出现时3个工作日内提交。
- 责任主体:NDA/ANDA申请人负责提交FAR,即使与第三方签订合同,仍承担最终责任。
- 提交时限:收到相关信息后3个工作日内必须提交FAR,逾期可能面临监管行动。
- 提交方式:推荐使用Form FDA 3331a电子形式提交FAR,加快审核流程。
- 后续与最终FAR:虽非强制,但建议提交,以便FDA评估公共卫生风险和公司回应的充分性。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |





{kind=link}