首页
>
资讯
>
国内药政每周导读:CDE 境外转境内化药申报资料,药典委新增大量通则等公示,参比目录公示第82批,上海局检查计划
出自识林
国内药政每周导读:CDE 境外转境内化药申报资料,药典委新增大量通则等公示,参比目录公示第82批,上海局检查计划
2024-05-13
【创新与临床】
5.6,【CDE】关于发布《中药新药用于紧张型头痛的临床疗效评价技术指导原则(试行)》的通告(2024年第21号)
本文曾于2023年9月征求意见 。
紧张型头痛是原发性头痛最常见的类型之一,典型特征为双侧轻中度压迫样或紧箍样头痛,部分患者伴有颅周压痛,日常活动后不加重。
紧张型头痛一般归属于中医内伤头痛、头风病的范畴。中医药对紧张型头痛的治疗,在减少发作频率、减轻疼痛程度、改善中医证候相关的其他症状或状态(如:焦虑状态、抑郁状态、失眠症状)等方面,具有治疗作用和独特优势。
【CMC药学研究】
5.6,【CDE】关于公开征求《化学仿制药参比制剂目录(第八十二批)》(征求意见稿)意见的通知
截至目前,NMPA已发布参比制剂 目录79批,CDE已公示82批。
识林用户可登录“中国参比制剂库 ”查阅。
5.10,【药典会】新增大量通则、辅料、生物制品和化药标准草案的公示
此次草案数量众多,且涉及到大量通用性的分析方法、剂型标准,乃至方法学验证,转移,实验室管理等等,例如(仅为部分):
识林会员可点击识林药典会标准公示数据库 ,查阅全部的最新药典标准公示。也可查阅“中国药典数据库 ”。
【审评与注册】
5.9,【CDE】关于发布《已上市境外生产药品转移至境内生产的药品上市注册申请申报资料要求(化学药品)》的通告(2024年第22号)
本文曾于2023年3月征求意见 。
该文的背景是前不久发布的《国家药监局关于优化已在境内上市的境外生产药品转移至境内生产的药品上市注册申请相关事项的公告 (2024年第49号)》 。
点击识林“花脸稿”可以看到,相对征求意见,改动之处不少,主要的倾向是要求更加简化,促进境外转境内,但也不乏更为明确的监管要求,例如:
删去“境外生产药品注册 或者再注册 时提交的药品生产国或者地区出具的药品批准证明文件复印件(如适用)”以及其他几个证明性文件。
规格方面,给出明确路径“对于同一药品存在多个规格 的,可进行部分规格转移,后续境内申请人如增加其他未转移规格,应按照已在境内上市的境外生产药品转移至境内生产的药品上市注册申请程序的要求提出相应申请。同一药品的不同规格应转移至同一境内申请人 。”
删去“转移后药品所使用的原料药,辅料种类、用量,一般不允许变更”的要求,而是“应提供与转移方原使用的原辅料 进行对比的资料”
对于原辅包登记,“转移至境内生产药品所用的原料药 、辅料 与直接接触药品的包装材料 和容器 参照常规仿制药要求执行。鼓励进行原料药登记,对于登记确有困难的,在所使用原料药供应商等与转移前完全一致的情况下,可在制剂中一并提交原料药相关资料。”
提出多个要求,如“关于转移次数。已上市境外生产药品不得按此路径多次转移至境内不同持有人生产(即境外 A 企业转移至境内 B 企业后,在 B 企业持有有效文号期间,不得再次由 A 企业转移至境内其他 C 企业)。”
建议RA阅读花脸稿,深入理解监管导向。
5.12,【CDE】药品审评报告和说明书更新,其中包括下列国产新药
注:仅列出国产创新药和改良型新药 ,多受理号仅展示其中1个,供参考。企业用户可至识林“CDE 药品审评报告”按发布时间查阅最新的上市药品信息。
受理号
药品名称
药品类型
注册分类 企业名称
承办日期
CXSS2200080
重组人促卵泡激素注射液
治疗用生物制品
3.4
长春金赛
2022-09-27
CXSS2200081
重组人促卵泡激素注射液
治疗用生物制品
3.4
长春金赛
2022-09-27
CXSS2200082
重组人促卵泡激素注射液
治疗用生物制品
3.4
长春金赛
2022-09-27
CXSS2200077
舒格利单抗注射液
治疗用生物制品
2.2
辉瑞
2022-09-08
CXHS2200039
奥雷巴替尼片
化药
2.4
广州顺健
2022-07-19
【GMP与检查】
5.10,【NMPA】关于恢复进口、销售和使用 Celgene Corporation 注射用紫杉醇(白蛋白结合型)的公告(2024年第60号)
被NMPA发了“警告信”的外企,花了4年时间,变更 了生产地址,终于得以再次上市。
根据药品境外生产现场检查结果,国家药监局于2020年3月25日发布公告(2020年 第44号) ,决定暂停进口、销售和使用Celgene Corporation注射用紫杉醇(白蛋白结合型)(英文名称:Paclitaxel For Injection (Albumin Bound),原注册证号:H20130650,受托生产企业:Fresenius Kabi USA, LLC,生产地址:2020 Ruby Street, Melrose Park, IL 60160, USA)。
Celgene Corporation向国家药监局提交注册补充申请和恢复进口、销售和使用的申请,将受托生产企业变更为Abraxis BioScience, LLC,生产地址为:620 N. 51st Avenue,Phoenix, AZ 85043 United States of America。国家药监局按照注册审评程序,经组织技术审查,已批准其变更受托生产企业。
5.11,【上海】2024年药品质量安全行政检查计划
本市辖区内药品上市许可持有人 、药品生产经营企业和使用单位(医疗机构);医疗机构制剂室;疫苗接种点;疫苗 储存配送企业;药品网络交易第三方平台;药用辅料和直接接触药品的包装材料包装材料和容器生产企业。必要时依法开展延伸检查。
以现场检查为主;根据检查工作实际需要,可以采取对被检查对象提交书面材料的检查、利用数字化技术手段开展远程检查等。
上海局还给出了基于风险的检查频率,例如:
对麻醉药品 、第一类精神药品 、药品类易制毒化学品生产企业每季度检查不少于一次,对第二类精神药品生产企业每半年检查不少于一次;
对疫苗 、血液制品 、放射性药品 、医疗用毒性药品、无菌药品 等高风险药品生产企业,每年不少于一次GMP 符合性检查;
对细胞治疗药品生产企业开展一次GMP符合性检查和一次日常监督检查;
结合上年度信用等级评估结果,对C级及以下企业开展一次GMP符合性检查和一次日常监督检查;
对非高风险药品生产企业,结合上年度信用等级评估结果,优化检查频次,对A级企业原则上可免于本年度日常监督检查(但3年内应开展一次日常监督检查、5年内开展一次GMP符合性检查)
其他类型企业的检查频率,请见原文。
【新药批准和报产】
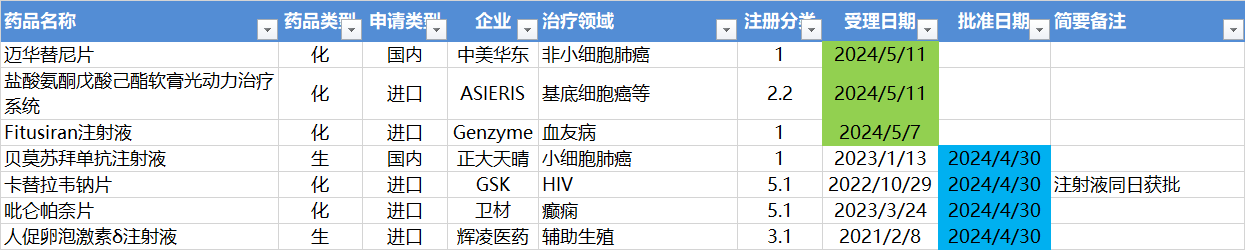
5.6-5.12,NMPA发布4个新药批准,CDE受理3个NDA
注:仅列出新药(包括改良型新药 )的上市申请和批准上市信息。在“以临床价值为导向”的背景下,申请上市以及获批的品种,其适应症 、临床研究 策略、注册路径,都值得业界关注和分析。
识林® 版权所有,未经许可不得转载
必读岗位及工作建议:
QA(质量保证):确保公司仿制药研发与生产过程符合参比制剂目录要求。 注册:关注参比制剂目录更新,及时调整注册策略。 研发:在药物开发过程中,以参比制剂为标杆,确保产品质量。 文件适用范围:
要点总结:
参比制剂目录更新 :强调了第八十二批化学仿制药参比制剂目录的公示和征求意见过程。反馈机制 :明确了通过参比制剂遴选申请平台进行反馈的方法,要求提供充分依据和论证材料。公示期限 :规定了公示期限为2024年5月6日至5月16日,共计10个工作日。未通过审议品种 :列举了未通过审议的品种及其原因,如缺乏足够的疗效和安全性数据。变更后上市许可持有人 :指出了部分品种变更后的上市许可持有人信息。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
注册专员 :熟悉新政策,准备和提交药品上市注册申请。研发部门 :了解转移生产的相关研究资料要求,确保研究符合规定。质量保证(QA) :监督药品生产转移过程中的质量控制。文件适用范围:
文件要点总结:
境内申请人责任 :已在境内上市的境外生产药品转移至境内生产,需由境内申请人按药品上市注册申请要求提出申请。原注册资料提交 :申请人可提交原境外生产药品的注册资料,并附加境内生产转移的相关研究资料。优先审评审批 :原研化学药品和生物制品转移至境内生产的注册申请将纳入优先审评审批范围。审评资料要求 :具体申报资料要求将由国家药监局药品审评中心另行制定发布。政策目的 :优化外商投资环境,促进医药行业高质量发展,提高药品可及性。以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。 QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA(质量保证):确保生产过程符合新修订的不溶性微粒检查法标准。 QC(质量控制):负责按照新标准执行不溶性微粒的检查工作。 R&D(研发):在研发阶段考虑不溶性微粒检查法对产品质量的影响。 适用范围说明:
要点总结:
标准修订目的 :强调修订《中国药典》0903不溶性微粒检查法,以提高标准的科学性、合理性和适用性。公示征求意见 :明确公示期为一个月,鼓励社会各界提出异议和建议。反馈要求 :规定了反馈意见的格式要求,包括加盖公章或个人签名,并提供联系方式。公示期满默认无异议 :若公示期满未收到反馈,则视为无异议。联系信息 :提供了联系人、电话、电子邮箱、通信地址等详细信息,方便意见反馈。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA:确保质量控制流程与新标准保持一致。 研发:调整药物研发过程中的溶出度与释放度测试方法。 注册:更新注册文件以符合新标准要求。 文件适用范围:
文件要点总结:
标准修订征求意见 :强调了对0931溶出度与释放度测定法的修订,并公开征求意见,以确保标准的科学性、合理性和适用性。公示期限与反馈机制 :明确公示期为30天,要求有异议的反馈需在线提交,并附有相关说明、实验数据和联系方式。反馈形式要求 :规定了反馈意见需打印并加盖公章或个人签名,以确保反馈的正式性和可追溯性。公示期满默认无异议 :指出公示期满后未回复意见即视为对标准草案无异议。联系信息提供 :提供了联系人、电话、电子邮箱和通信地址等详细信息,以便相关方进行咨询和反馈。以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA:应仔细研读公示稿,确保分析方法验证流程符合新标准。 研发:需关注分析方法验证的更新,以确保研发过程中方法的合规性。 注册:了解新标准对注册申报的影响,准备相应的注册资料。 适用范围:
要点总结:
公示目的 :强调了修订《中国药典》9101分析方法验证指导原则的重要性,以提高标准的科学性、合理性和适用性。意见征集 :明确公示期间为三个月,鼓励社会各界提出意见和建议,以确保标准的全面性和准确性。反馈要求 :规定了反馈意见的具体形式,包括加盖公章或个人签名,并提供了详细的联系方式。公示期限 :明确了公示期的起止日期,以及未回复意见即视为无异议的规定。联系信息 :提供了联系人、电话、电子邮箱和通信地址等详细信息,方便相关方进行沟通和反馈。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA:确保实验室质量管理符合新指导原则。 研发:了解微生物实验室质量管理的最新要求,指导研发流程。 生产:掌握微生物实验室操作规范,保证生产过程合规。 文件适用范围:
文件要点总结:
公示目的 :确保9203药品微生物实验室质量管理指导原则的科学性、合理性和适用性。公示期限 :公示期为60天,自2024年5月6日至7月6日。反馈要求 :若有异议,需在线反馈,并提供相关说明、实验数据和联系方式。反馈形式 :来函需加盖公章或个人签名,并邮寄至指定地址。默认同意 :公示期满未回复意见即视为对草案无异议。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA(质量保证):确保洁净实验室的微生物监测和控制符合新修订的指导原则。 QC(质量控制):负责实验室微生物监测的具体操作和数据记录。 研发:在新药研发过程中,需遵循微生物监测和控制的规范。 适用范围说明:
文件要点总结:
修订目的 :强调修订9205指导原则以确保其科学性、合理性和适用性。公示征求意见 :明确公示期为两个月,鼓励社会各界提出异议和建议。反馈要求 :规定反馈需包含相关说明、实验数据和联系方式,个人反馈需签名。公示标准草案异议处理 :公示期满未回复意见视为无异议。联系信息 :提供了联系人、电话、电子邮箱和通信地址等详细信息。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA(质量保证):确保制药用水的微生物监测和控制符合新指导原则。 QC(质量控制):执行微生物监测程序,确保数据准确性。 生产:遵循微生物控制措施,保证生产过程用水安全。 文件适用范围:
文件要点总结:
公示征求意见 :公示期为60天,鼓励社会各界提出异议和建议。微生物监测标准 :明确制药用水的微生物监测要求,确保水质安全。控制措施 :规定制药用水的微生物控制措施,防止微生物污染。反馈机制 :提供详细的反馈途径,包括在线反馈和邮寄反馈。公示期满默认同意 :公示期满未回复意见即视为对草案无异议。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA(质量保证):确保分析用天平与称量操作符合《中国药典》的新标准。 研发:在药品研发过程中,确保称量准确性,符合公示的指导原则。 生产:按照新标准调整生产过程中的称量操作,确保产品质量。 适用范围说明:
文件要点总结:
公示目的 :确保《中国药典》中分析用天平与称量指导原则的科学性、合理性和适用性。征求意见 :公示期为三个月,鼓励社会各界提出异议和建议。反馈要求 :反馈需详细说明、实验数据支持,并加盖公章或个人签名。公示期限 :公示期满未回复意见即视为无异议。联系信息 :提供了联系人、电话、电子邮箱和通信地址等详细信息。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA:确保质量控制流程符合微生物分析方法验证和确认要求。 研发:在新药开发过程中,遵循微生物分析方法的验证和转移指导原则。 生产:在生产过程中应用经过验证的微生物分析方法,确保产品质量。 文件适用范围:
文件要点总结:
方法验证要求: 明确了药品微生物分析方法必须经过严格的验证过程,以确保分析结果的准确性和可靠性。方法确认指导: 提供了方法确认的具体指导,确保分析方法适用于特定药品的微生物检测。方法转移规定: 强调了在不同实验室或生产场所间转移微生物分析方法时,必须遵循特定的程序和标准。公示征求意见: 公示草案以征求社会各界意见,确保指导原则的科学性和适用性。反馈机制: 设立了反馈机制,允许相关利益方提出异议并提供实验数据支持。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA:确保非无菌产品的质量控制符合新指导原则。 生产:了解并实施微生物风险控制措施。 研发:在产品设计阶段考虑微生物风险控制。 文件适用范围:
文件要点总结:
公示目的 :确保非无菌产品微生物风险控制标准的科学性、合理性和适用性。意见征集 :公示期为60天,鼓励社会各界提出异议和建议。反馈要求 :反馈需详细、附有实验数据,并提供联系方式。公示结果 :公示期满未回复意见即视为无异议。联系信息 :提供了联系人、电话、电子邮箱和通信地址。以上仅为部分要点,请阅读原文,深入理解监管要求。