首页
>
资讯
>
FDA 表示将彻底改革受到批评的医疗器械体系
出自识林
2019-01-04
美国FDA官员在2018年11月26日表示,计划彻底改革已经有数十年历史的大多数医疗器械审批的体系。专家们长期以来一直批评该这一体系没有发现有风险的植入物和医疗器械的问题。FDA宣布了旨在确保新医疗器械能够反映最新安全性和有效性的计划。改革所针对的510(k) 体系,通常允许器械生产商基于与几十年前产品的相似性推出新产品,而不是基于在患者中开展新的临床检验结果推出新产品。也就是说,新器械仍然必须具有与前基器械相同的预期用途,但新器械的技术特性可以与安全性和性能标准进行比较,而不是与前基器械的技术特性相比较。
记者调查披露,FDA辩驳
FDA宣布此举前一天的11月25日,包括50多家媒体机构对医疗器械安全进行全球调查发布。由国际调查记者同盟(International Consortium of Investigative Journalists,ICIJ)牵头,多家知名媒体和BMJ参与的这项调查发现,FDA在十年间收到医疗机构报告的怀疑与医疗器械有关的超过170万例伤害和近83,000例死亡事件。除了采访医生、患者、研究人员和公司内部举报者之外,记者还收集并分析了数以百万计的医疗记录、召回 通知和其它产品安全警告。
这项调查还发现,被其它国家视为医疗器械监管的黄金标准的FDA采取的简化审批流程的方式,让使用者处于危险之中,而在迫使公司纠正时有发生的危及生命的问题时,却反应缓慢。即便发生重大问题时,医疗器械也鲜有撤市。FDA并未披露每年在美国发生的医疗器械移植有多少例,这一数据对准确计算移植的成功率和失败率至关重要。
FDA承认其数据存在局限性,包括错误、遗漏和报告不足,因此很难确定器械是否直接造成伤害或死亡。但FDA拒绝了认为存在监督失灵的看法。“美国市场上有超过190,000种不同的器械。我们每个工作日批准或许可十几种新的或经过改进的器械,”FDA器械与放射健康中心(CDRH)主任Jeffrey Shuren博士在今年5月的一次行业会议上表示。“新闻界在任一时刻关注的少数几种器械,比我们在一个工作日内投放市场的器械要少。对我来说,并不意味着系统失灵。这一系统表现正常。”
记者调查同时曝光企业违规行为
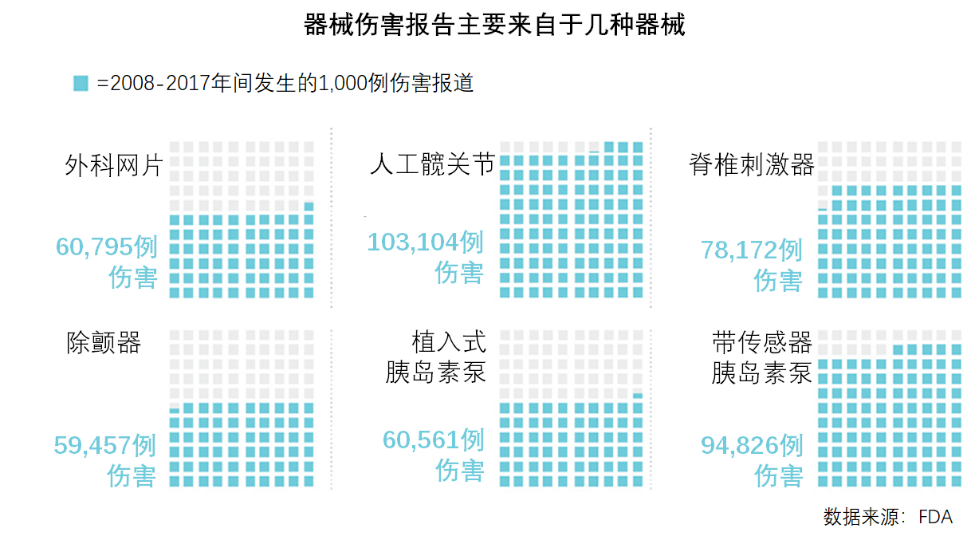
尽管FDA的器械伤害数据涉及约4,000类医疗器械,但自2008年以来,其中的6类器械所涉伤害就占了总伤害数的四分之一。每年移植到50,000-60,000位患者的脊椎刺激器引起的伤害,在所有涉及伤害的医疗器械中名列第3(下图)。
与此同时,医疗器械生产商也花费数十亿美元试图影响监管机构、医院和医生。在美国,药品和器械生产商需要披露给医生支付的款项。ICIJ组织分析医疗保险和医疗补助服务中心的数据表明,2017年,10家最大的医疗器械公司向医生或医院支付了近6亿美元,用于支付咨询费、研究费、旅行费和娱乐费。这一数字不包括强生公司和Allergan等也销售其它产品的器械生产商的付款。游说记录显示,自2017年以来,四大脊髓刺激器生产商已花费超过2200万美元,试图通过影响立法使其整体业务受益,其中也涵盖其它器械。
根据联合调查披露,一些公司因贿赂医生、非法推销未经批准用途的产品,支付宣称其产品安全性和有效性的研究而被处以罚款。2016年,美国最大的内窥镜和相关医疗器械分销商美国奥林巴斯公司同意向美国司法部支付6.232亿美元,“用于与向医生和医院支付回扣计划相关的刑事指控和民事索赔和解”,奥林巴斯公司表示“同意对其合规计划进行各种改进。” 2016年,司法部指控美敦力公司通过向医生提供具有强大的财务诱惑让医生变成推销员,伤害患者和欺诈联邦医疗保健计划,美敦力公司支付280万美元和解。
FDA局长:一代人以来的“最重要的现代化举措”
上世纪70年代,数以万计的女性受到达康盾(Dalkon Shield)宫内节育器引起的致命感染伤害。消费者权益组织强烈要求医疗器械上市前经过检测和上市前审批,防止有缺陷产品引起的死亡和伤害。1976年,国会通过《医疗器械修正案》(Medical Device Amendments),对《食品、药品和化妆品法案 》做出修订,首次要求要求FDA测试和审批包括宫内节育器在内的医疗器械。
为了回应记者提出的问题,FDA在今年11月表示,正在采取新行动,通过更好的数据为患者创建更强大的医疗器械安全网。但在器械进入市场之前,FDA不可能全方位了解一种器械的风险与获益。在过去的半个世纪,通过推出治疗或诊断心脏病,癌症和糖尿病的器械,医疗器械行业彻底改变了现代医学中一些最致命的疾病的治疗方法。
“我们认为现在是时候对1976年首次采用的方法进行根本性的现代化改革,” FDA局长Scott Gottlieb和器械与放射健康中心(CDRH)主任Jeffrey Shuren博士在2018年11月26日发布的一份声明中表示,正在考虑的改革将促使公司将其器械与更新的技术进行比较。Gottlieb在Twitter上将该提案描述为一个时代中医疗器械审评流程的“最重要的现代化举措”。
在这份声明中提出的改革要点包括:
(1)要求生产商在寻求通过510(k) 获批时使用更加现代的前基器械或客观的性能标准。FDA表示,目前510(k)获批的近20%是依据超过10年的前基器械。FDA认为,这意味着某些器械没有持续改进,但FDA是否会鼓励这种改进,在各种情况下需要的更现代的前基器械?FDA正在采取前瞻性的方法,而不是扮演被动的监管机构的角色。但仍然有理由使用与安全性无关的老旧的前基器械,例如在某类的器械没有快速发展的情况下。
(2)FDA正在考虑公开基于与超过10年的前基器械比较而获批的器械。FDA将寻求公众对这种方法的评论,包括是否应该考虑其它标准,以及是否有其它方法来促进使用更现代的前基器械。尽管FDA一再声明,依赖旧前基器械并不意味着器械不安全,但这样的做法会产生一种印象,即基于旧器械批准的器械有问题。否则,非专业人士可能会合理地询问,为什么要发布列表?这样的做法,更多的是出于支持基于市场的考虑而不是出于政府干预。也就是说,如果患者和医生更喜欢基于更现代的前基器械的器械,那么基于老旧前基器械的器械的市场将会缩减。
(3)FDA计划在2019年早些时候最终敲定2018年4月公布的《扩展简化510(k)计划指南草案:通过性能标准证明实质性等效 》(Expansion of the Abbreviated 510(k) Program: Demonstrating Substantial Equivalence through Performance Criteria)。今年4月公布的《医疗器械安全行动计划:保护患者,促进公众健康》(Medical Device Safety Action Plan:Protecting Patients, Promoting Public Health )中,FDA表示允许器械生产商使用客观的性能标准来证明实质等效性(而不是与前基器械比较)可能会推动更大的市场竞争,开发更为安全的器械。同月发布的《扩展简化510(k)计划指南草案》进一步证明了FDA鼓励使用客观性能标准而非前基器械的计划。今年11月26日发布的声明,消除了对于FDA愿景的任何不确定性:新途径被设想为“从技术上取代生产商将器械与特定的,有时是老旧的前基器械进行比较的做法。”“从技术上”这一提法很重要,在不改变法律的情况下,新器械仍必须具有与前基器械相同的预期用途,但新器械的技术特性可以与安全性和性能标准进行比较,而不是前基器械的技术特性。
FDA提出的一些改革,可能需要数年时间才能实施,可能包括针对生产商的新指南和法规,而最具实质性的变化可能需要国会采取行动。
FDA审评体系牵头专家David Challoner博士表示,这些变化可能会提高器械安全性,但担心生产商可能会阻挠改革工作。认为如果器械行业通过游说力图重归原样,将可能会导致这样的改革半途而废。2011年,Challoner领导的一个国立医学研究院(IOM)顾问小组建议取代FDA “存在缺陷 ”的审评制度。当时,FDA表示不同意该小组的建议。
长期以来,政府监管机构和独立医学专家的报告一直批评在FDA美国市场上批准95%以上器械所倚赖的架构。与要求在患者研究中检测的新药不同,大多数医疗器械只需要表明它与市场上已有的器械相似。只有少数真正的新器械必须经过广泛的临床检验验证是否安全有效。
Challoner的评审小组得出的结论是,国会最初的目的是简化通道,成为当时市场上数以千计的风险从低度到中度的“祖父”级器械的临时通道。但这样的通道并没有被逐步淘汰,反而成为主要的审批途径。通过简化体系批准的有缺陷的器械,包括未到寿就提前失效的髋关节置换装置,与疼痛和出血相关的外科网片,以及意外传播子宫癌的手术器械。
随着多代器械都通过FDA的主要审评流程获批,医疗产品变得越来越复杂,通常已经几乎变得不像这些器械生产商所声称参照的几十年前的“前基器械”(predicate device)。通过这一被称为510(k)体系获批的器械,包括成像扫描仪、电脑输液泵、人工关节和脊椎内移植物。
FDA表示,将考虑逐步淘汰一些老旧器械,使得参照这些器械的新产品无法进入市场。FDA指出,通过510(k)审核流程获批的器械中,有近20%是基于超过10年的前基器械。
行业组织:期待对FDA的计划有更多了解
医疗器械行业的主要游说组织先进医疗技术协会(The Advanced Medical Technology Association,AdvaMed)在一份声明中表示,期待对FDA的计划有更多了解。
该组织表示,尽管多年来已证明510(k)路径的有效性,但一直认为任何流程都有能够加以改进的余地。协会中负责技术和监管事务的Janet Trunzo表示,医疗器械公司在资本和人力方面投入了无数资源,开发前沿的合规计划。
作者:识林-Kapok® 版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
参考资料 Statement from FDA Commissioner Scott Gottlieb, M.D. and Jeff Shuren, M.D., Director of the Center for Devices and Radiological Health, on transformative new steps to modernize FDA's 510(k) program to advance the review of the safety and effectiveness of medical devices. Nov 26, 2018. https://www.icij.org/investigations/implant-files/ https://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM604195.pdf https://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDRH/CDRHReports/UCM604690.pdf